
Genetic Background of Congenital Erythrocytosis


Abstract
:1. Introduction
2. Primary Congenital Erythrocytosis
2.1. EPOR
2.2. LNK/SH2B3
3. Secondary Congenital Erythrocytosis
3.1. The Oxygen Sensing Pathway
3.2. VHL
3.3. PHD2/EGLN1
3.4. HIF2A/EPAS1
3.5. ERYTHROPOIETIN Gene
3.6. High Oxygen Affinity Variants
3.7. Methemoglobinemia
3.8. Bisphosphoglycerate Mutase Deficiency
3.9. PIEZO1
3.10. SLC30A10 Mutations with Hypermanganesemia
4. Management of Congenital Erythrocytosis
5. Idiopathic Erythrocytosis
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pearson, T.C.; Guthrie, D.L.; Simpson, J.; Chinn, S.; Barosi, G.; Ferrant, A.; Lewis, S.M.; Najean, Y. Interpretation of measured red cell mass and plasma volume in adults: Expert Panel on Radionuclides of the International Council for Standardization in Haematology. Br. J. Haematol. 1995, 89, 748–756. [Google Scholar] [CrossRef]
- Johansson, P.L.; Safai-Kutti, S.; Kutti, J. An elevated venous haemoglobin concentration cannot be used as a surrogate marker for absolute erythrocytosis: A study of patients with polycythaemia vera and apparent polycythaemia. Br. J. Haematol. 2005, 129, 701–705. [Google Scholar] [CrossRef]
- De la Chapelle, A.; Träskelin, A.L.; Juvonen, E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc. Natl. Acad. Sci. USA 1993, 90, 4495–4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percy, M.J.; McMullin, M.F.; Roques, A.W.; Westwood, N.B.; Acharya, J.; Hughes, A.E.; Lappin, T.R.; Pearson, T.C. Erythrocytosis due to a mutation in the erythropoietin receptor gene. Br. J. Haematol. 1998, 100, 407–410. [Google Scholar] [CrossRef]
- Bento, C.; Percy, M.J.; Gardie, B.; Maia, T.M.; van Wijk, R.; Perrotta, S.; Della Ragione, F.; Almeida, H.; Rossi, C.; Girodon, F.; et al. ECE-Consortium, Genetic basis of congenital erythrocytosis: Mutation update and online databases. Hum. Mutat. 2014, 35, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, F.; Marty, C.; Balligand, T.; Verdier, F.; Grosjean, S.; Gryshkova, V.; Raslova, H.; Constantinescu, S.N.; Casadevall, N.; Vainchenker, W.; et al. New pathogenic mechanisms induced by germline erythropoietin receptor mutations in primary erythrocytosis. Haematologica 2018, 103, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef]
- McMullin, M.F. HIF pathway mutations and erythrocytosis. Expert Rev. Hematol. 2010, 3, 93–101. [Google Scholar] [CrossRef]
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621. [Google Scholar] [CrossRef]
- Gordeuk, V.R.; Sergueeva, A.I.; Miasnikova, G.Y.; Okhotin, D.; Voloshin, Y.; Choyke, P.L.; Butman, J.A.; Jedlickova, K.; Prchal, J.T.; Polyakova, L.A. Congenital disorder of oxygen sensing: Association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood 2004, 103, 3924–3932. [Google Scholar] [CrossRef]
- Percy, M.J.; McMullin, M.F.; Jowitt, S.N.; Potter, M.; Treacy, M.; Watson, W.H.; Lappin, T.R. Chuvash-type congenital polycythemia in 4 families of Asian and Western European ancestry. Blood 2003, 102, 1097–1099. [Google Scholar] [CrossRef] [Green Version]
- Perrotta, S.; Nobili, B.; Ferraro, M.; Migliaccio, C.; Borriello, A.; Cucciolla, V.; Martinelli, V.; Rossi, F.; Punzo, F.; Cirillo, P.; et al. Von Hippel-Lindau-dependent polycythemia is endemic on the island of Ischia: Identification of a novel cluster. Blood 2006, 107, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Percy, M.J.; Amos, C.I.; Guan, Y.; Shete, S.; Stockton, D.W.; McMullin, M.F.; Polyakova, L.A.; Ang, S.O.; Pastore, Y.D.; et al. The worldwide distribution of the VHL 598C>T mutation indicates a single founding event. Blood 2004, 103, 1937–1940. [Google Scholar] [CrossRef]
- Lenglet, M.; Robriquet, F.; Schwarz, K.; Camps, C.; Couturier, A.; Hoogewijs, D.; Buffet, A.; Knight, S.J.L.; Gad, S.; Couvé, S.; et al. Identification of a new. Blood 2018, 132, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percy, M.J.; Zhao, Q.; Flores, A.; Harrison, C.; Lappin, T.R.; Maxwell, P.H.; McMullin, M.F.; Lee, F.S. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; Furlow, P.W.; Beer, P.A.; Lappin, T.R.; McMullin, M.F.; Lee, F.S. A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood 2007, 110, 2193–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, R.; Syed, N.; Shah, P. Erythrocytosis due to PHD2 Mutations: A Review of Clinical Presentation, Diagnosis, and Genetics. Case Rep. Hematol. 2016, 2016, 6373706. [Google Scholar]
- Arsenault, P.R.; Pei, F.; Lee, R.; Kerestes, H.; Percy, M.J.; Keith, B.; Simon, M.C.; Lappin, T.R.J.; Khurana, T.S.; Lee, F.S. A knock-in mouse model of human PHD2 gene-associated erythrocytosis establishes a haploinsufficiency mechanism. J. Biol. Chem. 2013, 288, 33571–33584. [Google Scholar] [CrossRef] [Green Version]
- Sinnema, M.; Song, D.; Guan, W.; Janssen, J.W.H.; van Wijk, R.; Navalsky, B.E.; Peng, K.; Donker, A.E.; Stegmann, A.P.A.; Lee, F.S. Loss-of-function zinc finger mutation in the. Blood 2018, 132, 1455–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladroue, C.; Carcenac, R.; Leporrier, M.; Gad, S.; Le Hello, C.; Galateau-Salle, F.; Feunteun, J.; Pouysségur, J.; Richard, S.; Gardie, B. PHD2 mutation and congenital erythrocytosis with paraganglioma. N. Engl. J. Med. 2008, 359, 2685–2692. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhuang, Z.; Fliedner, S.M.; Shankavaram, U.; Sun, M.G.; Bullova, P.; Zhu, R.; Elkahloun, A.G.; Kourlas, P.J.; Merino, M.; et al. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J. Mol. Med. 2015, 93, 93–104. [Google Scholar] [CrossRef]
- Percy, M.J.; Beer, P.A.; Campbell, G.; Dekker, A.W.; Green, A.R.; Oscier, D.; Rainey, M.G.; van Wijk, R.; Wood, M.; Lappin, T.R.; et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood 2008, 111, 5400–5402. [Google Scholar] [CrossRef]
- Tan, Q.; Kerestes, H.; Percy, M.J.; Pietrofesa, R.; Chen, L.; Khurana, T.S.; Christofidou-Solomidou, M.; Lappin, T.R.; Lee, F.S. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J. Biol. Chem. 2013, 288, 17134–17144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, D.P.; Harten, S.K.; Reid, C.D.; Tuddenham, E.G.; Maxwell, P.H. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 2008, 112, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Yang, C.; Lorenzo, F.; Merino, M.; Fojo, T.; Kebebew, E.; Popovic, V.; Stratakis, C.A.; Prchal, J.T.; Pacak, K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N. Engl. J. Med. 2012, 367, 922–930. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, F.R.; Yang, C.; Ng Tang Fui, M.; Vankayalapati, H.; Zhuang, Z.; Huynh, T.; Grossmann, M.; Pacak, K.; Prchal, J.T. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J. Mol. Med. 2013, 91, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Zmajkovic, J.; Lundberg, P.; Nienhold, R.; Torgersen, M.L.; Sundan, A.; Waage, A.; Skoda, R.C. A Gain-of-Function Mutation in EPO in Familial Erythrocytosis. N. Engl. J. Med. 2018, 378, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Charache, S.; Weatherall, D.J.; Clegg, J.B. Polycythemia associated with a hemoglobinopathy. J. Clin. Investig. 1966, 45, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; Butt, N.N.; Crotty, G.M.; Drummond, M.W.; Harrison, C.; Jones, G.L.; Turner, M.; Wallis, J.; McMullin, M.F. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Haematologica 2009, 94, 1321–1322. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; McFerran, N.V.; Lappin, T.R. Disorders of oxidised haemoglobin. Blood Rev. 2005, 19, 61–68. [Google Scholar] [CrossRef]
- Petousi, N.; Copley, R.R.; Lappin, T.R.; Haggan, S.E.; Bento, C.M.; Cario, H.; Percy, M.J.; Ratcliffe, P.J.; Robbins, P.A.; McMullin, M.F.; et al. Erythrocytosis associated with a novel missense mutation in the BPGM gene. Haematologica 2014, 99, e201–e204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazana, I.; Mohamedali, A.; Smith, F.; de Lavallade, H.; McLornan, D.; Raj, K. Uniparental disomy (UPD) of a novel bisphosphoglycerate mutase (BPGM) mutation leading to erythrocytosis. Br. J. Haematol. 2021, 192, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Picard, V.; Guitton, C.; Thuret, I.; Rose, C.; Bendelac, L.; Ghazal, K.; Aguilar-Martinez, P.; Badens, C.; Barro, C.; Bénéteau, C.; et al. Clinical and biological features in. Haematologica 2019, 104, 1554–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filser, M.; Giansily-Blaizot, M.; Grenier, M.; Monedero Alonso, D.; Bouyer, G.; Pérès, L.; Egée, S.; Aral, B.; Airaud, F.; Da Costa, L.; et al. Increased incidence of germline PIEZO1 mutations in individuals with idiopathic erythrocytosis. Blood 2021, 137, 1828–1832. [Google Scholar] [CrossRef] [PubMed]
- Tuschl, K.; Clayton, P.T.; Gospe, S.M.; Gulab, S.; Ibrahim, S.; Singhi, P.; Aulakh, R.; Ribeiro, R.T.; Barsottini, O.G.; Zaki, M.S.; et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 2012, 90, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergueeva, A.I.; Miasnikova, G.Y.; Polyakova, L.A.; Nouraie, M.; Prchal, J.T.; Gordeuk, V.R. Complications in children and adolescents with Chuvash polycythemia. Blood 2015, 125, 414–415. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.G.; Brooks, J.T.; Balanos, G.M.; Lappin, T.R.; Layton, D.M.; Leedham, D.L.; Liu, C.; Maxwell, P.H.; McMullin, M.F.; McNamara, C.J.; et al. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006, 3, e290. [Google Scholar] [CrossRef] [Green Version]
- Formenti, F.; Beer, P.A.; Croft, Q.P.; Dorrington, K.L.; Gale, D.P.; Lappin, T.R.; Lucas, G.S.; Maher, E.R.; Maxwell, P.H.; McMullin, M.F.; et al. Cardiopulmonary function in two human disorders of the hypoxia-inducible factor (HIF) pathway: Von Hippel-Lindau disease and HIF-2alpha gain-of-function mutation. FASEB J. 2011, 25, 2001–2011. [Google Scholar] [CrossRef]
- McMullin, M.F.F.; Mead, A.J.; Ali, S.; Cargo, C.; Chen, F.; Ewing, J.; Garg, M.; Godfrey, A.; Knapper, S.; McLornan, D.P.; et al. A guideline for the management of specific situations in polycythaemia vera and secondary erythrocytosis: A British Society for Haematology Guideline. Br. J. Haematol. 2019, 184, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Landolfi, R.; Marchioli, R.; Kutti, J.; Gisslinger, H.; Tognoni, G.; Patrono, C.; Barbui, T.; For the European Collaboration on Low-Dose Aspirin in Polycythemia Vera Investigators. Efficacy and safety of low-dose aspirin in polycythemia vera. N. Engl. J. Med. 2004, 350, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.C.; Sufan, R.I.; Zhou, B.; Heir, P.; Bunda, S.; Sybingco, S.S.; Greer, S.N.; Roche, O.; Heathcote, S.A.; Chow, V.W.; et al. Loss of JAK2 regulation via a heterodimeric VHL-SOCS1 E3 ubiquitin ligase underlies Chuvash polycythemia. Nat. Med. 2011, 17, 845–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, A.W.; Knoche, E.M.; Engle, E.K.; Ban-Hoefen, M.; Kaiwar, C.; Oh, S.T. Clinical Improvement with JAK2 Inhibition in Chuvash Polycythemia. N. Engl. J. Med. 2016, 375, 494–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, E.; Cho, K.H.; Harris, S.T.; Flindt, N.R.; Watt, R.K.; Pai, A.B. Hypoxia-inducible factor prolyl hydroxylase inhibitors: A paradigm shift for treatment of anemia in chronic kidney disease? Expert Opin. Investig. Drugs 2020, 29, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Bauer, T.M.; Papadopoulos, K.P.; Plimack, E.R.; Merchan, J.R.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Perini, R.F.; et al. Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: A phase 1 trial and biomarker analysis. Nat. Med. 2021, 27, 802–805. [Google Scholar] [CrossRef]
- Camps, C.; Petousi, N.; Bento, C.; Cario, H.; Copley, R.R.; McMullin, M.F.; van Wijk, R.; Ratcliffe, P.J.; Robbins, P.A.; Taylor, J.C.; et al. Gene panel sequencing improves the diagnostic work-up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica 2016, 101, 1306–1318. [Google Scholar] [CrossRef]
- Biagetti, G.; Catherwood, M.; Robson, N.; Bertozzi, I.; Cosi, E.; McMullin, M.F.; Randi, M.L. HFE mutations in idiopathic erythrocytosis. Br. J. Haematol. 2018, 181, 270–272. [Google Scholar] [CrossRef] [Green Version]
- Burlet, B.; Bourgeois, V.; Buriller, C.; Aral, B.; Airaud, F.; Garrec, C.; Bézieau, S.; Gardie, B.; Girodon, F. High HFE mutation incidence in idiopathic erythrocytosis. Br. J. Haematol. 2019, 185, 794–795. [Google Scholar] [CrossRef] [Green Version]


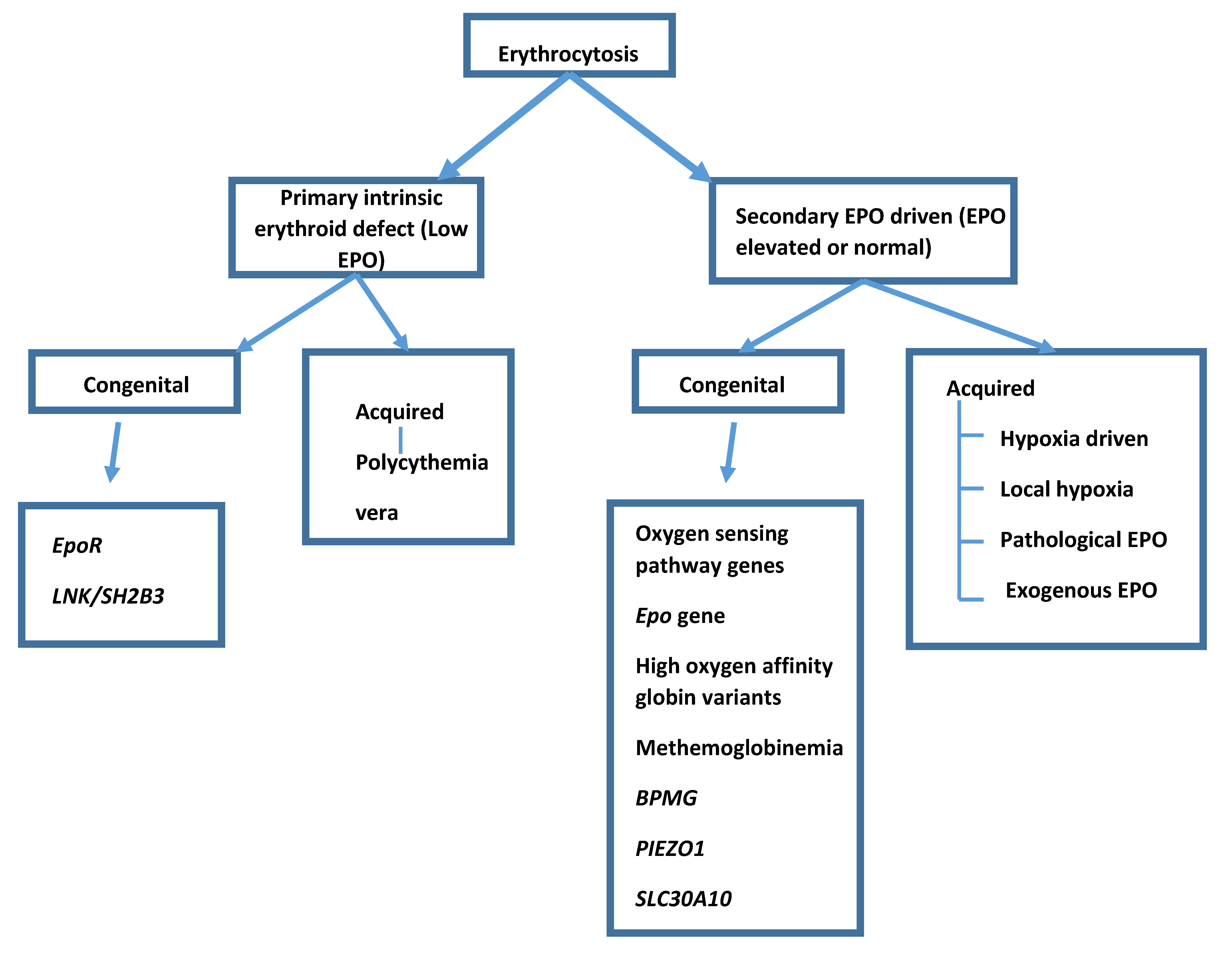{kind=link}
| Primary | ||
| Erythropoietin receptor LNK/SH2B3 | ||
| Secondary | ||
| Oxygen sensing pathway genes | ||
| VHL PHD2/EGLN1 HIF2A/EPAS1 | ||
| Erythropoietin gene | ||
| α and β globin genes (High oxygen affinity Hbs) Methemoglobinemia (Abnormal M Hb or cytochrome reductase deficiency) BPMG PIEZ01 SLC30A10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McMullin, M.F. Genetic Background of Congenital Erythrocytosis. Genes 2021, 12, 1151. https://doi.org/10.3390/genes12081151
McMullin MF. Genetic Background of Congenital Erythrocytosis. Genes. 2021; 12(8):1151. https://doi.org/10.3390/genes12081151
Chicago/Turabian StyleMcMullin, Mary Frances. 2021. "Genetic Background of Congenital Erythrocytosis" Genes 12, no. 8: 1151. https://doi.org/10.3390/genes12081151
APA StyleMcMullin, M. F. (2021). Genetic Background of Congenital Erythrocytosis. Genes, 12(8), 1151. https://doi.org/10.3390/genes12081151