
Rett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review


Abstract
:1. Introduction
2. Typical RTT
2.1. Epilepsy
2.2. Movement Disorder (MD) and Stereotypies
2.3. Genotype-Phenotype Correlations
3. RTT Variants
3.1. From “Early-Onset Seizure” Variant to “CDKL5-Related” Disorder
3.2. Epilepsy
3.3. From Congenital RTT Variant to FOXG1 Syndrome
3.4. Epilepsy
3.5. MD and Stereotypies
4. RETT-Like Phenotypes
4.1. Syndromic Conditions
4.1.1. Pitt–Hopkins Syndrome (TCF4) and Pitt-Hopkins-Like Syndrome (CNTNAP2, NRXN1)
4.1.2. Cornelia de Lange (CdL) Syndrome (SMC1A, HDAC8, NIPBL, SMC3, RAD21)
4.1.3. Phelan–McDermid Syndrome (SHANK3)
4.1.4. Christianson Type X-Linked Mental Retardation Syndrome (SLC9A6, NHE6)
4.1.5. Glass Syndrome (SATB2)
4.1.6. HNRNPU-Related Disorders
4.1.7. MEIS2
5. Monogenic Non-Syndromic Conditions
5.1. DE/EE Genes
5.2. STXBP1
5.3. Ion Channels
5.3.1. SCN1A
5.3.2. SCN2A
5.3.3. SCN8A
5.3.4. KCNB1
5.3.5. KCNQ2
5.3.6. HCN1
5.4. Receptors
5.4.1. GABRB3
5.4.2. GABRG2
5.4.3. GRIA2
5.4.4. GRIN1
5.4.5. GRIN2B
5.5. Transporters
5.5.1. SLC6A1
5.5.2. SLC35A2
5.6. Transcription Factors
5.6.1. MEF2C
5.6.2. ACTL6B
5.7. Axon Guidance
NTNG1
5.8. Ubiquitination
5.8.1. RHOBTB2
5.8.2. HECW2
5.9. Intellectual Disability and Epilepsy Genes
5.9.1. Synapsis
IQSEC2
5.9.2. Transcription Regulation/Modification
HNRNPH2
EEF1A2
5.9.3. Neurodegenerative Disorders
WDR45
5.9.4. PPT1
MFSD8
EIF2B2
6. Epilepsy and DE/EE Genes with Stereotypies
7. Clinical Remarks
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [Green Version]
- Schönewolf-Greulich, B.; Bisgaard, A.-M.; Møller, R.; Dunø, M.; Brøndum-Nielsen, K.; Kaur, S.; Van Bergen, N.; Lunke, S.; Eggers, S.; Jespersgaard, C.; et al. Clinician’s guide to genes associated with Rett-like phenotypes-Investigation of a Danish cohort and review of the literature. Clin. Genet. 2018, 95, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Pintaudi, M.; Calevo, M.G.; Vignoli, A.; Parodi, E.; Aiello, F.; Baglietto, M.G.; Hayek, J.; Buoni, S.; Renieri, A.; Russo, S.; et al. Epilepsy in Rett syndrome: Clinical and genetic features. Epilepsy Behav. 2010, 19, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Frullanti, E.; Papa, F.T.; Grillo, E.; Clarke, A.; Ben-Zeev, B.; Pineda, M.; Bahi-Buisson, N.; Bienvenu, T.; Armstrong, J.; Martinez, A.R.; et al. Analysis of the Phenotypes in the Rett Networked Database. Int. J. Genom. 2019, 2019, 6956934–6956939. [Google Scholar] [CrossRef]
- Nissenkorn, A.; Levy-Drummer, R.S.; Bondi, O.; Renieri, A.; Villard, L.; Mari, F.; Mencarelli, M.A.; Rizzo, C.L.; Meloni, I.; Pineda, M.; et al. Epilepsy in Rett syndrome-Lessons from the Rett networked database. Epilepsia 2015, 56, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Hagne, I.; Witt-Engerstrom, I.; Hagberg, B. EEG development in Rett syndrome. A study of 30 cases. Electroencephalogr. Clin. Neurophysiol. 1989, 72, 1–6. [Google Scholar] [CrossRef]
- Seltzer, L.E.; Ma, M.; Ahmed, S.; Bertrand, M.; Dobyns, W.B.; Wheless, J.; Paciorkowski, A.R. Epilepsy and outcome inFOXG1-related disorders. Epilepsia 2014, 55, 1292–1300. [Google Scholar] [CrossRef] [Green Version]
- Cellini, E.; Vignoli, A.; Pisano, T.; Falchi, M.; Molinaro, A.; Accorsi, P.; Bontacchio, A.; Pinelli, L.; Giordano, L.; Guerrini, R. The hyperkinetic movement disorder of FOXG1-related epileptic-dyskinetic encephalopathy. Dev. Med. Child Neurol. 2015, 58, 93–97. [Google Scholar] [CrossRef]
- Lopes, F.; Barbosa, M.; Ameur, A.; Soares, G.; de Sá, J.; Dias, A.I.; Oliveira, G.; Cabral, P.; Temudo, T.; Calado, E.; et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 2016, 53, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, Q.; Chen, Y.; Yu, S.; Wu, X.; Bao, X. Rett and Rett-like syndrome: Expanding the genetic spectrum to KIF1A and GRIN1 gene. Mol. Genet. Genom. Med. 2019, 7, e968. [Google Scholar] [CrossRef] [Green Version]
- Cogliati, F.; Giorgini, V.; Masciadri, M.; Bonati, M.T.; Marchi, M.; Cracco, I.; Gentilini, D.; Peron, A.; Savini, M.N.; Spaccini, L.; et al. Pathogenic Variants in STXBP1 and in Genes for GABAa Receptor Subunities Cause Atypical Rett/Rett-like Phenotypes. Int. J. Mol. Sci. 2019, 20, 3621. [Google Scholar] [CrossRef] [Green Version]
- Sáez, M.A.; Fernández-Rodríguez, J.; Moutinho, C.; Sanchez-Mut, J.V.; Gomez, A.; Vidal, E.; Petazzi, P.; Szczesna, K.; López-Serra, P.; Lucariello, M.; et al. Mutations in JMJD1C are involved in Rett syndrome and intellectual disability. Genet. Med. 2015, 18, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M.; et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131, 2647–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahi-Buisson, N.; Kaminska, A.; Boddaert, N.; Rio, M.; Afenjar, A.; Gérard, M.; Giuliano, F.; Motte, J.; Héron, D.; Morel, M.A.N.; et al. The three stages of epilepsy in patients withCDKL5mutations. Epilepsia 2008, 49, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Parrini, E. Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia 2012, 53, 2067–2078. [Google Scholar] [CrossRef]
- Melani, F.; Mei, D.; Pisano, T.; Savasta, S.; Franzoni, E.; Ferrari, A.R.; Marini, C.; Guerrini, R. CDKL5 gene-related epileptic encephalopathy: Electroclinical findings in the first year of life. Dev. Med. Child Neurol. 2011, 53, 354–360. [Google Scholar] [CrossRef]
- Kortüm, F.; Das, S.; Flindt, M.; Morris-Rosendahl, D.; Stefanova, I.; Goldstein, A.; Horn, D.; Klopocki, E.; Kluger, G.; Martin, P.; et al. The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J. Med. Genet. 2011, 48, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Vegas, N.; Cavallin, M.; Maillard, C.; Boddaert, N.; Toulouse, J.; Schaefer, E.; Lerman-Sagie, T.; Lev, D.; Magalie, B.; Moutton, S.; et al. Delineating FOXG1 syndrome: From congenital microcephaly to hyperkinetic encephalopathy. Neurol. Genet. 2018, 4, e281. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, A.; Schneider, R.B.; Augustine, E.F.; Ng, J.; Mankad, K.; Meyer, E.; McTague, A.; Ngoh, A.; Hemingway, C.; Robinson, R.; et al. Delineation of the movement disorders associated with FOXG1 mutations. Neurology 2016, 86, 1794–1800. [Google Scholar] [CrossRef] [Green Version]
- Ehrhart, F.; Sangani, N.B.; Curfs, L.M. Current developments in the genetics of Rett and Rett-like syndrome. Curr. Opin. Psychiatry 2018, 31, 103–108. [Google Scholar] [CrossRef]
- Temudo, T.; Santos, M.; Dias, K.; Calado, E.; Carrilho, I.; Oliveira, G.G.; Barbot, C.; Fonseca, M.; Cabral, A.; Dias, A.; et al. Stereotypies in Rett syndrome: Analysis of 83 patients with and without detected MECP2 mutations. Neurology 2007, 68, 1183–1187. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Paciorkowski, A.R.; Ciccone, R.; Della Mina, E.; Bonaglia, M.C.; Borgatti, R.; Schaaf, C.P.; Sutton, V.R.; Xia, Z.; Jelluma, N.; et al. Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur. J. Hum. Genet. 2010, 19, 102–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milh, M.; Villeneuve, N.; Chouchane, M.; Kaminska, A.; Laroche, C.; Barthez, M.A.; Gitiaux, C.; Bartoli, C.; Borges-Correia, A.; Cacciagli, P.; et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia 2011, 52, 1828–1834. [Google Scholar] [CrossRef]
- Larsen, J.; Carvill, G.L.; Gardella, E.; Kluger, G.; Schmiedel, G.; Barisic, N.; Depienne, C.; Brilstra, E.; Mang, Y.; Nielsen, J.E.K.; et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015, 84, 480–489. [Google Scholar] [CrossRef] [Green Version]
- Ohba, C.; Kato, M.; Takahashi, S.; Lerman-Sagie, T.; Lev, D.; Terashima, H.; Kubota, M.; Kawawaki, H.; Matsufuji, M.; Kojima, Y.; et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 2014, 55, 994–1000. [Google Scholar] [CrossRef]
- Yoo, Y.; Jung, J.; Lee, Y.-N.; Lee, Y.; Cho, H.; Na Bs, E.; Hong, J.; Kim, E.; Lee, J.S.; Lee, J.S.; et al. GABBR2mutations determine phenotype in rett syndrome and epileptic encephalopathy. Ann. Neurol. 2017, 82, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.E.; Tambunan, D.; LaCoursiere, C.; Goldenberg, M.; Pinsky, R.; Martin, E.; Ho, E.; Khwaja, O.; Kaufmann, W.E.; Poduri, A. Mutations in epilepsy and intellectual disability genes in patients with features of Rett syndrome. Am. J. Med. Genet. A 2015, 167, 2017–2025. [Google Scholar] [CrossRef] [Green Version]
- Allou, L.; Julia, S.; Amsallem, D.; El Chehadeh, S.; Lambert, L.; Thevenon, J.; Duffourd, Y.; Saunier, A.; Bouquet, P.; Pere, S.; et al. Rett-like phenotypes: Expanding the genetic heterogeneity to the KCNA2 gene and first familial case of CDKL5-related disease. Clin. Genet. 2016, 91, 431–440. [Google Scholar] [CrossRef]
- Romaniello, R.; Saettini, F.; Panzeri, E.; Arrigoni, F.; Bassi, M.T.; Borgatti, R. A de-novo STXBP1 gene mutation in a patient showing the Rett syndrome phenotype. NeuroReport 2015, 26, 254–257. [Google Scholar] [CrossRef]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.-M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes and genotypes in individuals with SMC1A variants. Am. J. Med. Genet. A 2017, 173, 2108–2125. [Google Scholar] [CrossRef] [Green Version]
- Lebrun, N.; Lebon, S.; Jeannet, P.-Y.; Jacquemont, S.; Billuart, P.; Bienvenu, T. Early-onset encephalopathy with epilepsy associated with a novel splice site mutation inSMC1A. Am. J. Med. Genet. A 2015, 167, 3076–3081. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.H.; Tim-Aroon, T.; Shieh, J.; Merrill, M.; Deeb, K.K.; Zhang, S.; Bass, N.E.; Bedoyan, J.K. Novel SMC1A frameshift mutations in children with developmental delay and epilepsy. Eur. J. Med. Genet. 2015, 58, 562–568. [Google Scholar] [CrossRef]
- Saikusa, T.; Hara, M.; Iwama, K.; Yuge, K.; Ohba, C.; Okada, J.-I.; Hisano, T.; Yamashita, Y.; Okamoto, N.; Saitsu, H.; et al. De novo HDAC8 mutation causes Rett-related disorder with distinctive facial features and multiple congenital anomalies. Brain Dev. 2018, 40, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Ohba, C.; Yamashita, Y.; Saitsu, H.; Matsumoto, N.; Matsuishi, T. De novoSHANK3mutation causes Rett syndrome-like phenotype in a female patient. Am. J. Med. Genet. A 2015, 167, 1593–1596. [Google Scholar] [CrossRef]
- Sajan, S.A.; Jhangiani, S.N.; Muzny, D.M.; Gibbs, R.A.; Lupski, J.R.; Glaze, D.G.; Kaufmann, W.E.; Skinner, S.A.; Annese, F.; Friez, M.J.; et al. Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet. Med. 2016, 19, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucariello, M.; Vidal, E.; Vidal, S.; Saez, M.; Roa, L.; Huertas, D.; Pineda, M.; Dalfó, E.; Dopazo, J.; Jurado, P.; et al. Whole exome sequencing of Rett syndrome-like patients reveals the mutational diversity of the clinical phenotype. Qual. Life Res. 2016, 135, 1343–1354. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Yoo, Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J. SATB2-associated syndrome presenting with Rett-like phenotypes. Clin. Genet. 2016, 89, 728–732. [Google Scholar] [CrossRef]
- LeDuc, M.S.; Chao, H.-T.; Qu, C.; Walkiewicz, M.; Xiao, R.; Magoulas, P.; Pan, S.; Beuten, J.; He, W.; Bernstein, J.A.; et al. Clinical and molecular characterization of de novo loss of function variants inHNRNPU. Am. J. Med. Genet. A 2017, 173, 2680–2689. [Google Scholar] [CrossRef]
- Shimada, S.; Oguni, H.; Otani, Y.; Nishikawa, A.; Ito, S.; Eto, K.; Nakazawa, T.; Yamamoto-Shimojima, K.; Takanashi, J.-I.; Nagata, S.; et al. An episode of acute encephalopathy with biphasic seizures and late reduced diffusion followed by hemiplegia and intractable epilepsy observed in a patient with a novel frameshift mutation in HNRNPU. Brain Dev. 2018, 40, 813–818. [Google Scholar] [CrossRef]
- Spagnoli, C.; Rizzi, S.; Salerno, G.G.; Frattini, D.; Koskenvuo, J.; Fusco, C. Pharmacological Treatment of Severe Breathing Abnormalities in a Case of HNRNPU Epileptic Encephalopathy. Mol. Syndromol. 2021, 12, 101–105. [Google Scholar] [CrossRef]
- Srivastava, S.; Desai, S.; Cohen, J.; Smith-Hicks, C.; Barañano, K.; Fatemi, A.; Naidu, S. Monogenic disorders that mimic the phenotype of Rett syndrome. Neurogenetics 2018, 19, 41–47. [Google Scholar] [CrossRef]
- Vidal, S.; Xiol, C.; Pascual-Alonso, A.; O’Callaghan, M.; Pineda, M.; Armstrong, J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. Int. J. Mol. Sci. 2019, 20, 3925. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.-S.; Lin, L.-J.; Yang, M.-T.; Wang, J.-S.; Lu, J.-F. The therapeutic implication of a novel SCN2A mutation associated early-onset epileptic encephalopathy with Rett-like features. Brain Dev. 2017, 39, 877–881. [Google Scholar] [CrossRef]
- Mastrangelo, M.; Manti, F.; Giannini, M.T.; Guerrini, R.; Leuzzi, V. KCNQ2 encephalopathy manifesting with Rett-like features: A follow-up into adulthood. Neurol. Genet. 2020, 6, e510. [Google Scholar] [CrossRef] [PubMed]
- Spagnoli, C.; Salerno, G.G.; Frattini, D.; Fusco, C. Two Cases of KCNQ2 Encephalopathy with Unusual Findings: Clinical and Neurophysiological Follow-Up. Clin. Pediatr. 2018, 1, 1001. [Google Scholar]
- Møller, R.S.; Wuttke, T.V.; Helbig, I.; Marini, C.; Johannesen, K.M.; Brilstra, E.H.; Vaher, U.; Borggraefe, I.; Talvik, I.; Talvik, T.; et al. Mutations in GABRB3: From febrile seizures to epileptic encephalopathies. Neurology 2017, 88, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; SYNAPS Study Group; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kyriakopoulos, P.; McNiven, V.; Carter, M.T.; Humphreys, P.; Dyment, D.; Fantaneanu, T.A. Atypical Rett Syndrome and Intractable Epilepsy with Novel GRIN2B Mutation. Child Neurol. Open 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienvenu, T.; Diebold, B.; Chelly, J.; Isidor, B. Refining the phenotype associated with MEF2C point mutations. Neurogenetics 2012, 14, 71–75. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Q.; Chen, Y.; Yu, S.; Wu, X.; Bao, X.; Wen, Y. Novel MEF2C point mutations in Chinese patients with Rett (−like) syndrome or non-syndromic intellectual disability: Insights into genotype-phenotype correlation. BMC Med. Genet. 2018, 19, 191. [Google Scholar] [CrossRef]
- Rocha, H.; Sampaio, M.; Rocha, R.; Fernandes, S.; Leão, M. MEF2C haploinsufficiency syndrome: Report of a new MEF2C mutation and review. Eur. J. Med. Genet. 2016, 59, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Rousseau, J.; Peng, H.; Aouabed, Z.; Priam, P.; Theroux, J.-F.; Jefri, M.; Tanti, A.; Wu, H.; Kolobova, I.; et al. Mutations in ACTL6B Cause Neurodevelopmental Deficits and Epilepsy and Lead to Loss of Dendrites in Human Neurons. Am. J. Hum. Genet. 2019, 104, 815–834. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Uematsu, M.; Numata-Uematsu, Y.; Abe, Y.; Endo, W.; Kikuchi, A.; Takezawa, Y.; Funayama, R.; Shirota, M.; Nakayama, K.; et al. Rett-like features and cortical visual impairment in a Japanese patient with HECW2 mutation. Brain Dev. 2018, 40, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Borg, I.; Freude, K.; Kübart, S.; Hoffmann, K.; Menzel, C.; Laccone, F.; Firth, H.V.; A Ferguson-Smith, M.; Tommerup, N.; Ropers, H.-H.; et al. Disruption of Netrin G1 by a balanced chromosome translocation in a girl with Rett syndrome. Eur. J. Hum. Genet. 2005, 13, 921–927. [Google Scholar] [CrossRef] [Green Version]
- Gandomi, S.K.; Gonzalez, K.D.F.; Parra, M.; Shahmirzadi, L.; Mancuso, J.; Pichurin, P.; Temme, R.; Dugan, S.; Zeng, W.; Tang, S. Diagnostic Exome Sequencing Identifies Two Novel IQSEC2 Mutations Associated with X-Linked Intellectual Disability with Seizures: Implications for Genetic Counseling and Clinical Diagnosis. J. Genet. Couns. 2013, 23, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Radley, J.A.; O’Sullivan, R.B.; Turton, S.E.; Cox, H.; Vogt, J.; Morton, J.; Jones, E.; Smithson, S.; Lachlan, K.; Rankin, J.; et al. Deep phenotyping of 14 new patients with IQSEC2 variants, including monozygotic twins of discordant phenotype. Clin. Genet. 2019, 95, 496–506. [Google Scholar] [CrossRef]
- Mau-Them, F.T.; Willems, M.; Albrecht, B.; Sanchez, E.; Puechberty, J.; Endele, S.; Schneider, A.; Pallares, N.R.; Missirian, C.; Rivier, F.; et al. Expanding the phenotype of IQSEC2 mutations: Truncating mutations in severe intellectual disability. Eur. J. Hum. Genet. 2013, 22, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Lopergolo, D.; Privitera, F.; Castello, G.; Lo Rizzo, C.; Mencarelli, M.A.; Pinto, A.M.; Ariani, F.; Currò, A.; Lamacchia, V.; Canitano, R.; et al. IQSEC2 disorder: A new disease entity or a Rett spectrum continuum? Clin. Genet. 2021, 99, 462–474. [Google Scholar] [CrossRef]
- Barrie, E.S.; Cottrell, C.E.; Gastier-Foster, J.; Hickey, S.E.; Patel, A.D.; Santoro, S.L.; Alfaro, M.P. Genotype-phenotype correlation: Inheritance and variant-type infer pathogenicity in IQSEC2 gene. Eur. J. Med. Genet. 2020, 63, 103735. [Google Scholar] [CrossRef]
- Howell, K.B.; McMahon, J.M.; Carvill, G.L.; Tambunan, D.; Mackay, M.T.; Rodriguez-Casero, V.; Webster, R.; Clark, D.; Freeman, J.L.; Calvert, S.; et al. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology 2015, 85, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Marini, C.; Romoli, M.; Parrini, E.; Costa, C.; Mei, D.; Mari, F.; Parmeggiani, L.; Procopio, E.; Metitieri, T.; Cellini, E.; et al. Clinical features and outcome of 6 new patients carrying de novo KCNB1 gene mutations. Neurol. Genet. 2017, 3, e206. [Google Scholar] [CrossRef] [Green Version]
- Saitsu, H.; Akita, T.; Tohyama, J.; Goldberg-Stern, H.; Kobayashi, Y.; Cohen, R.; Kato, M.; Ohba, C.; Miyatake, S.; Tsurusaki, Y.; et al. De novo KCNB1 mutations in infantile epilepsy inhibit repetitive neuronal firing. Sci. Rep. 2015, 5, 15199. [Google Scholar] [CrossRef] [Green Version]
- Nava, C.; Dalle, C.; Rastetter, A.; Striano, P.; De Kovel, C.G.F.; Nabbout, R.; Cancès, C.; Ville, D.; Brilstra, E.H.; EuroEPINOMICS RES Consortium; et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat. Genet. 2014, 46, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Pironti, E.; Granata, F.; Cucinotta, F.; Gagliano, A.; Efthymiou, S.; Houlden, H.; Salpietro, V.; Di Rosa, G. Electroclinical history of a five-year-old girl with GRIN1-related early-onset epileptic encephalopathy: A video-case study. Epileptic Disord. 2018, 20, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; McMahon, J.M.; Schneider, A.; Zemel, M.; Myers, C.T.; Saykally, J.; Nguyen, J.; Robbiano, A.; Zara, F.; Specchio, N.; et al. Mutations in the GABA Transporter SLC6A1 Cause Epilepsy with Myoclonic-Atonic Seizures. Am. J. Hum. Genet. 2015, 96, 808–815. [Google Scholar] [CrossRef] [Green Version]
- Yates, T.M.; Suri, M.; Desurkar, A.; Lesca, G.; Wallgren-Pettersson, C.; Hammer, T.B.; Raghavan, A.; Poulat, A.-L.; Møller, R.; Thuresson, A.-C.; et al. SLC35A2-related congenital disorder of glycosylation: Defining the phenotype. Eur. J. Paediatr. Neurol. 2018, 22, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, L.; Bienvenu, T.; Allou, L.; Valduga, M.; Echenne, B.; Diebold, B.; Mignot, C.; Héron, D.; Roth, V.; Saunier, A.; et al. MEF2C mutations are a rare cause of Rett or severe Rett-like encephalopathies. Clin. Genet. 2012, 82, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Nectoux, J.; Girard, B.; Bahi-Buisson, N.; Prieur, F.; Afenjar, A.; Rosas-Vargas, H.; Chelly, J.; Bienvenu, T. Netrin G1 Mutations Are an Uncommon Cause of Atypical Rett Syndrome with or Without Epilepsy. Pediatr. Neurol. 2007, 37, 270–274. [Google Scholar] [CrossRef]
- Straub, J.; Konrad, E.D.; Grüner, J.; Toutain, A.; Bok, L.A.; Cho, M.T.; Crawford, H.P.; Dubbs, H.; Douglas, G.; Jobling, R.; et al. Missense Variants in RHOBTB2 Cause a Developmental and Epileptic Encephalopathy in Humans, and Altered Levels Cause Neurological Defects in Drosophila. Am. J. Hum. Genet. 2018, 102, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Spagnoli, C.; Soliani, L.; Caraffi, S.G.; Baga, M.; Rizzi, S.; Salerno, G.G.; Frattini, D.; Garavelli, L.; Koskenvuo, J.; Pisani, F.; et al. Paroxysmal movement disorder with response to carbamazepine in a patient with RHOBTB2 developmental and epileptic encephalopathy. Park. Relat. Disord. 2020, 76, 54–55. [Google Scholar] [CrossRef]
- Bain, J.M.; Cho, M.T.; Telegrafi, A.; Wilson, A.; Brooks, S.; Botti, C.; Gowans, G.; Autullo, L.A.; Krishnamurthy, V.; Willing, M.C.; et al. Variants in HNRNPH2 on the X Chromosome Are Associated with a Neurodevelopmental Disorder in Females. Am. J. Hum. Genet. 2016, 99, 728–734. [Google Scholar] [CrossRef] [Green Version]
- Peron, A.; Novara, F.; La Briola, F.; Merati, E.; Giannusa, E.; Segalini, E.; Anniballi, G.; Vignoli, A.; Ciccone, R.; Canevini, M.P. Missense variants in the Arg206 residue of HNRNPH2: Further evidence of causality and expansion of the phenotype. Am. J. Med. Genet. A 2020, 182, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Kruer, M.C.; Gregory, A.; Haack, T.B.; Kurian, M.A.; Houlden, H.H.; Anderson, J.; Boddaert, N.; Sanford, L.; Harik, S.I.; et al. β-Propeller protein-associated neurodegeneration: A new X-linked dominant disorder with brain iron accumulation. Brain 2013, 136, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Belohlavkova, A.; Sterbova, K.; Betzler, C.; Burkhard, S.; Panzer, A.; Wolff, M.; Lassuthova, P.; Vlckova, M.; Kyncl, M.; Benova, B.; et al. Clinical features and blood iron metabolism markers in children with β-propeller protein associated neurodegeneration. Eur. J. Paediatr. Neurol. 2020, 28, 81–88. [Google Scholar] [CrossRef]
- Ohba, C.; Nabatame, S.; Iijima, Y.; Nishiyama, K.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Tanaka, F.; Ozono, K.; Saitsu, H.; et al. De novo WDR45 mutation in a patient showing clinically Rett syndrome with childhood iron deposition in brain. J. Hum. Genet. 2014, 59, 292–295. [Google Scholar] [CrossRef]
- Chard, M.; Appendino, J.P.; Bello-Espinosa, L.E.; Curtis, C.; Rho, J.M.; Wei, X.-C.; Al-Hertani, W. Single-center experience with β-propeller protein-associated neurodegeneration (BPAN); expanding the phenotypic spectrum. Mol. Genet. Metab. Rep. 2019, 20, 100483. [Google Scholar] [CrossRef] [PubMed]
- Hoffjan, S.; Ibisler, A.; Tschentscher, A.; Dekomien, G.; Bidinost, C.; Rosa, A.L. WDR45 mutations in Rett (-like) syndrome and developmental delay: Case report and an appraisal of the literature. Mol. Cell. Probes 2016, 30, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Ikeda, T.; Hasegawa, T.; Yamamoto, Y.; Kawato, K.; Komoto, T.; Imoto, I. Early manifestations of BPAN in a pediatric patient. Am. J. Med. Genet. A 2014, 164, 3095–3099. [Google Scholar] [CrossRef]
- Kulikovskaja, L.; Sarajlija, A.; Savic-Pavicevic, D.; Dobricic, V.; Klein, C.; Westenberger, A. WDR45 mutations may cause a MECP2 mutation-negative Rett syndrome phenotype. Neurol. Genet. 2018, 4, e227. [Google Scholar] [CrossRef] [Green Version]
- Topçu, M.; Tan, H.; Yalnizoğlu, D.; Usubutun, A.; Saatçi, I.; Aynaci, M.; Anlar, B.; Topaloğlu, H.; Turanli, G.; Köse, G.; et al. Evaluation of 36 patients from Turkey with neuronal ceroid lipofuscinosis: Clinical, neurophysiological, neuroradiological and histopathologic studies. Turk. J. Pediatr. 2004, 46, 1–10. [Google Scholar]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H. Major Facilitator Superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craiu, D.; Dragostin, O.; Dica, A.; Hoffman-Zacharska, D.; Gos, M.; Bastian, A.E.; Gherghiceanu, M.; Rolfs, A.; Nahavandi, N.; Craiu, M.; et al. Rett-like onset in late-infantile neuronal ceroid lipofuscinosis (CLN7) caused by compound heterozygous mutation in the MFSD8 gene and review of the literature data on clinical onset signs. Eur. J. Paediatr. Neurol. 2015, 19, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Kozina, A.A.; Okuneva, E.G.; Baryshnikova, N.V.; Krasnenko, A.Y.; Tsukanov, K.Y.; Klimchuk, O.I.; Kondakova, O.; Larionova, A.N.; Батышева, T.T.; Surkova, E.I.; et al. A novel MFSD8 mutation in a Russian patient with neuronal ceroid lipofuscinosis type 7: A case report. BMC Med. Genet. 2018, 19, 151. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yoo, Y.; Lim, B.C.; Kim, K.J.; Song, J.; Choi, M.; Chae, J.-H. GM3 synthase deficiency due toST3GAL5variants in two Korean female siblings: Masquerading as Rett syndrome-like phenotype. Am. J. Med. Genet. A 2016, 170, 2200–2205. [Google Scholar] [CrossRef]
- Jurecka, A.; Zikanova, M.; Tylki-Szymanska, A.; Krijt, J.; Bogdanska, A.; Gradowska, W.; Mullerova, K.; Sykut-Cegielska, J.; Kmoch, S.; Pronicka, E. Clinical, biochemical and molecular findings in seven Polish patients with adenylosuccinate lyase deficiency. Mol. Genet. Metab. 2008, 94, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Macchiaiolo, M.; Buonuomo, P.S.; Mastrogiorgio, G.; Bordi, M.; Testa, M.B.C.; Weber, G.; Bellacchio, E.; Tartaglia, M.; Cecconi, F.; Bartuli, A. Very mild isolated intellectual disability caused by adenylosuccinate lyase deficiency: A new phenotype. Mol. Genet. Metab. Rep. 2020, 23, 100592. [Google Scholar] [CrossRef]
- Kodera, H.; Ohba, C.; Kato, M.; Maeda, T.; Araki, K.; Tajima, D.; Matsuo, M.; Hino-Fukuyo, N.; Kohashi, K.; Ishiyama, A.; et al. De novoGABRA1mutations in Ohtahara and West syndromes. Epilepsia 2016, 57, 566–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sands, T.T.; Miceli, F.; Lesca, G.; Beck, A.E.; Sadleir, L.G.; Arrington, D.K.; Schönewolf-Greulich, B.; Moutton, S.; Lauritano, A.; Nappi, P.; et al. Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann. Neurol. 2019, 86, 181–192. [Google Scholar] [CrossRef]
- Smith, L.; Singhal, N.; El Achkar, C.M.; Truglio, G.; Sheidley, B.R.; Sullivan, J.; Poduri, A. PCDH19 -related epilepsy is associated with a broad neurodevelopmental spectrum. Epilepsia 2018, 59, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Tohyama, J.; Kato, M.; Akasaka, N.; Magara, S.; Kawashima, H.; Ohashi, T.; Shiraishi, H.; Nakashima, M.; Saitsu, H.; et al. High prevalence of genetic alterations in early-onset epileptic encephalopathies associated with infantile movement disorders. Brain Dev. 2016, 38, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Assoum, M.; Philippe, C.; Isidor, B.; Perrin, L.; Makrythanasis, P.; Sondheimer, N.; Paris, C.; Douglas, J.; Lesca, G.; Antonarakis, S.; et al. Autosomal-Recessive Mutations in AP3B2, Adaptor-Related Protein Complex 3 β 2 Subunit, Cause an Early-Onset Epileptic Encephalopathy with Optic Atrophy. Am. J. Hum. Genet. 2016, 99, 1368–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, K.M.; Meyer, E.; Grozeva, D.; Spinelli, E.; McTague, A.; Sanchis-Juan, A.; Carss, K.J.; Bryant, E.; Reich, A.; Schneider, A.L.; et al. Bi-allelic Loss-of-Function CACNA1B Mutations in Progressive Epilepsy-Dyskinesia. Am. J. Hum. Genet. 2019, 104, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Danti, F.R.; Galosi, S.; Romani, M.; Montomoli, M.; Carss, K.J.; Raymond, F.L.; Parrini, E.; Bianchini, C.; McShane, T.; Dale, R.C.; et al. GNAO1 encephalopathy: Broadening the phenotype and evaluating treatment and outcome. Neurol. Genet. 2017, 3, e143. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, C.; Angle, B.; Millichap, J. Early-life epileptic encephalopathy secondary to SZT2 pathogenic recessive variants. Epileptic Disord. 2016, 18, 195–200. [Google Scholar] [CrossRef]
- Vidal, S.; Brandi, N.; Pacheco, P.; Gerotina, E.; Blasco, L.; Trotta, J.R.; Derdak, S.; Del Mar O’Callaghan, M.; Garcia-Cazorla, À.; Pineda, M.; et al. The utility of Next Generation Sequencing for molecular diagnostics in Rett syndrome. Sci. Rep. 2017, 7, 12288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandweiss, A.J.; Brandt, V.L.; Zoghbi, H.Y. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: Prospects for future therapies. Lancet Neurol. 2020, 19, 689–698. [Google Scholar] [CrossRef]
- Gillentine, M.A.; Wang, T.; Hoekzema, K.; Rosenfeld, J.; Liu, P.; Guo, H.; Kim, C.N.; De Vries, B.B.A.; Vissers, L.E.L.M.; Nordenskjold, M.; et al. Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders. Genome Med. 2021, 13, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, F.; Coort, S.L.; Eijssen, L.; Cirillo, E.; Smeets, E.E.; Sangani, N.B.; Evelo, C.T.; Curfs, L.M.G. Integrated analysis of human transcriptome data for Rett syndrome finds a network of involved genes. World J. Biol. Psychiatry 2020, 21, 712–725. [Google Scholar] [CrossRef]




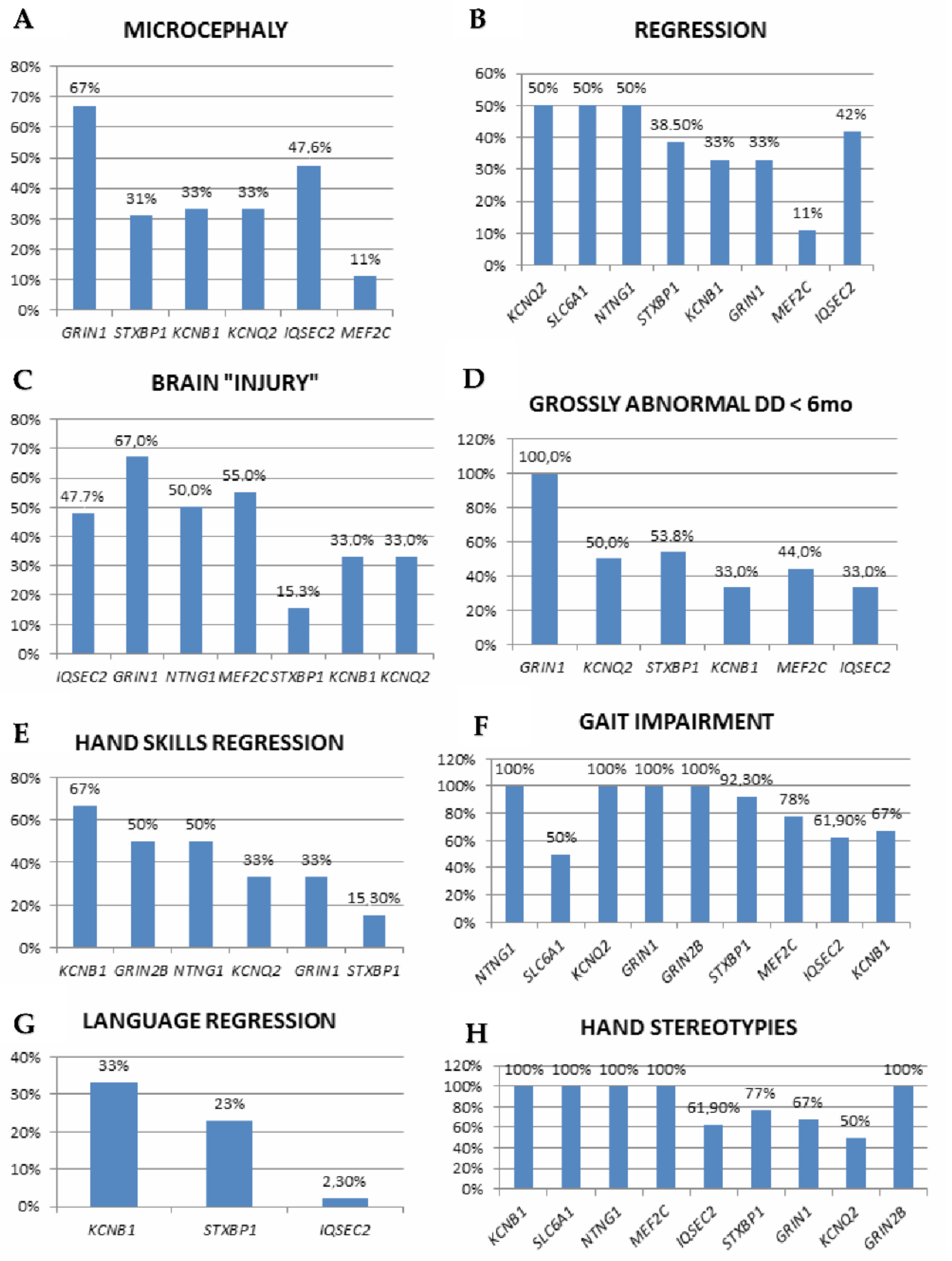{kind=link}
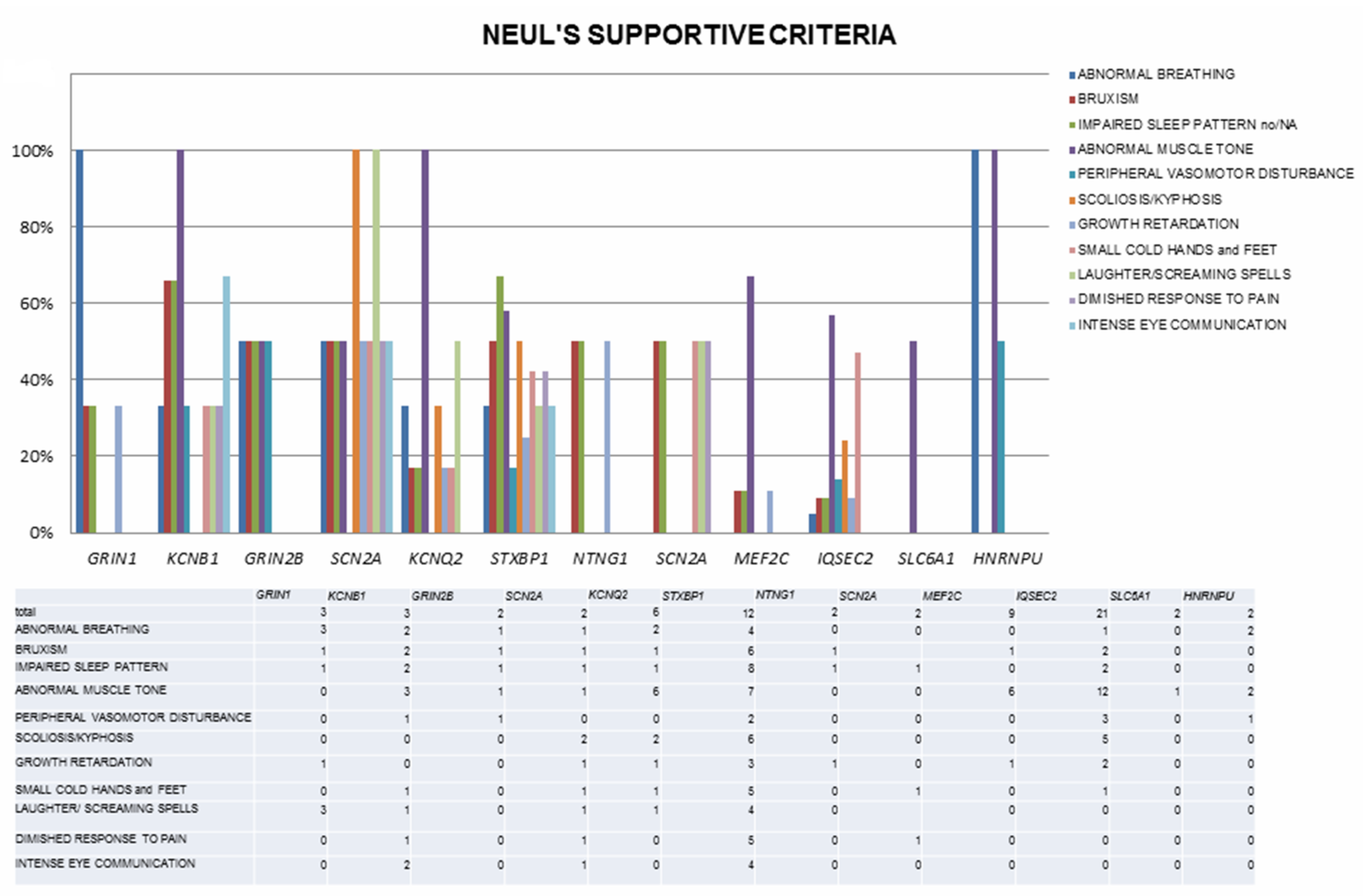{kind=link}
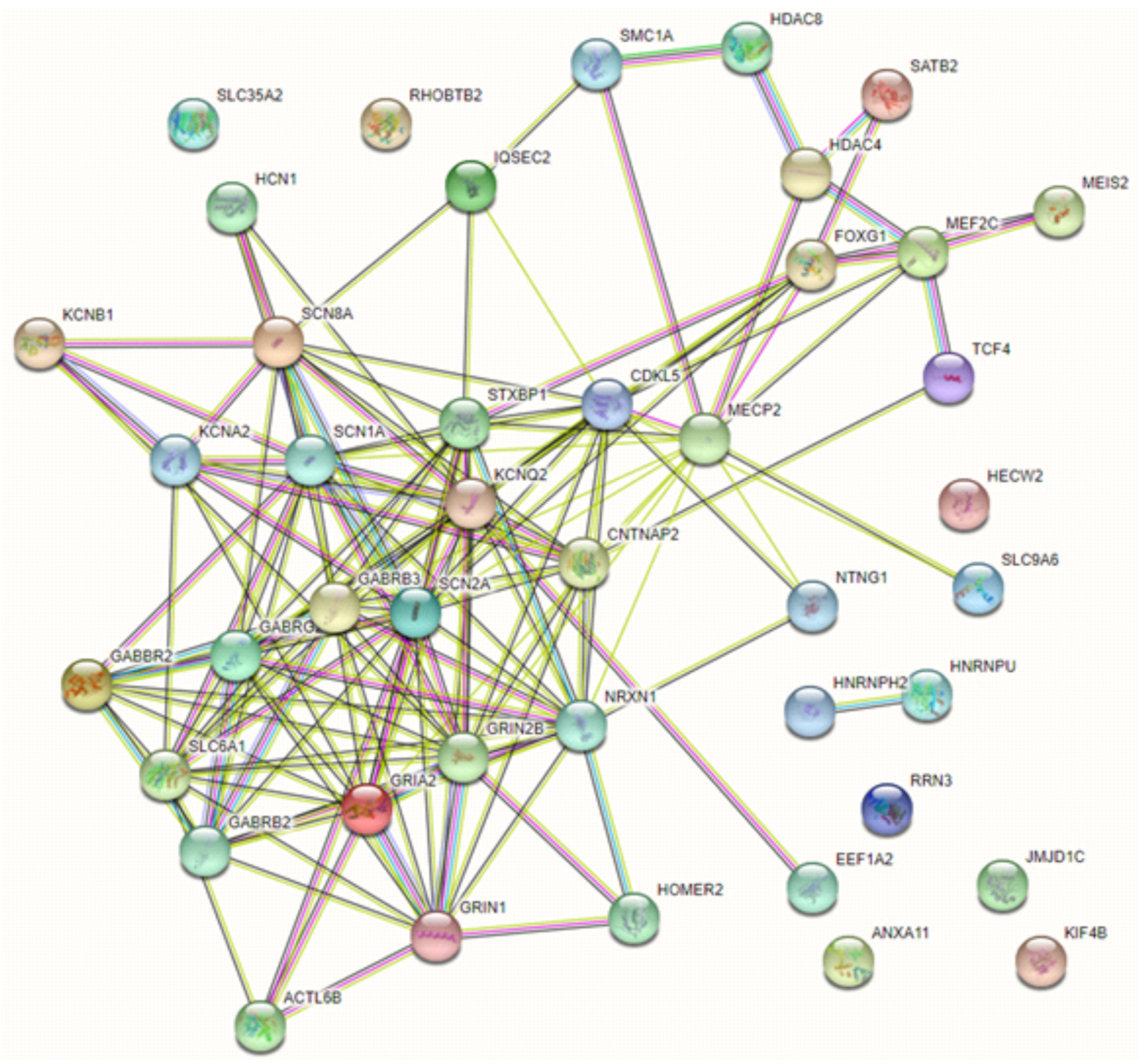{kind=link}
| Required for Typical RTT | Required for Atypical (Variant) RTT | Main Criteria | Exclusion Criteria for Typical RTT | Supportive Criteria for Atypical RTT |
|---|---|---|---|---|
|
|
|
|
|
| Typical RTT | CDKL5-Related Disorder | FOXG1 Syndrome |
|---|---|---|
| Clinical Stages | Electroclinical Stages | Epilepsy Features |
| Early onset phase (6–12 months): loss of acquired motor and language skills and purposeful hand movements Rapid destructive phase (1–3 y): autistic features, intellectual disability, hand stereotypies, abnormal gait/motor dysfunction, onset of abnormal respiratory patterns Stabilization phase (2–10 y): improvements in behavior, eye contact and hand function Late motor deterioration (>10 y): spasticity, dystonia, and scoliosis, loss of independent walking in ambulant patients EPILEPSY FEATURES Mean onset: 4.7 y Frequent FS, No specific seizure semiology | Stage I (Early epilepsy): IS; Tonic-vibratory seizure, followed by a clonic phase with series of spasms, gradually evolving into repetitive distal myoclonic jerks, lasting 2–4 min Onset: neonatal-4th month (median: 4 weeks) Stage II (EE): 6 months–3 years (median: 11 months) IS intermixed with brief tonic seizures profound DD, no language or motor development, massive hypotonia Stage III (Late multifocal and myoclonic epilepsy): ages: 2.5–11 y (median 7 y) drug-resistant epilepsy with tonic seizures and spasms, myoclonic jerks or atypical absences Or epilepsy remission Ages: 2.5–19 y (median 5 y) | Deletions and intragenic variants: epilepsy onset within the second year of life (mean: 22 months in [7]) various epilepsy types (focal impaired awareness, myoclonic, and bilateral tonic) rate of drug resistance is high Duplications: IS (mean age at onset: 7.4 months). Frequent focal seizures (onset: 5 months–6 years), often in association with spasms [8]. in a minority: later recurrence of tonic or myoclonic seizures |
| EEG | EEG | EEG: |
| Stage 1: N/posterior rhythms slowing Stage 2: rolandic IED (drowsiness, sleep). sleep architecture abnormalities (poor/absent spindles) Stage 3: abnormal background (posterior slowing, absent sleep figures); bilaterally synchronous bursts of pseudo-periodic delta and generalized rhythmic spikes in sleep Stage 4: abnormal, slow background (wakefulness and sleep), central and/or vertex theta (4–6 Hz), IED (multifocal spikes or sharp waves during wakefulness and generalized slow spike-wave complexes during sleep) | Stage I: Interictal: N/slow. Ictal: bilateral and synchronous flattening, followed by repetitive sharp waves and spikes Stage II: Typical/modified hypsarrhythmia, very slow, intermixed with focal spikes and polyspikes (F, C, O) Stage III: High-amplitude delta with pseudo-periodic bursts of high-amplitude S, PS, SW predominating over the C, T, T-O region | Deletions and intragenic variants: slow background, multifocal S and sharp waves, less frequently diffuse theta excess Duplications: hypsarrhythmia/modified hypsarrhythmia (onset) multifocal S-slowW, bursts of generalized S-slowW, or focal slowing intermixed with high-amplitude irregular S-slowW (follow-up) |
| GENE, Function/Name | Associated Disorder/OMIM# | Inheritance | Diagnosis within RSS Spectrum | REF |
|---|---|---|---|---|
| DE/EE Genes | ||||
| Synapsis | ||||
| Synaptic Vesicle Cycle | ||||
| STXBP1 | DEE4 (#612164) | AD | Atypical: 7/12 RTT-like: 4/12 “Typical”: 1/12 | [2,9,11,27,29,35] |
| Ion Channels | ||||
| SCN1A | Dravet syndrome (#607208) GEFS+ 2 (#604403) | AD | RTT-like (single case) | [36] |
| SCN2A | DEE11 (#613721) BFIS3 (#607745) | AD | Atypical: 1/2 RTT-like: 1/2 | [2,43] |
| SCN8A | DEE13(#614558) BFIS5 (#617080) Cognitive impairment w/out cerebellar ataxia (#614306) | AD | RTT-like (single case) | [27] |
| KCNB1 | DEE26 (#616056) | AD | Typical: 1/3 Atypical: 2/3 | [2,41] |
| KCNQ2 | DEE7 (#613720) Myokymia (#121200) BNS1 (#121200) | AD | RTT-like: 6/6 | [10,42,44,45] |
| HCN1 | DEE24 (#615871) GEFS+10 (#618482) | AD | RTT-like (single case) | [36] |
| KCNA2 | DEE32 (#616366) | AD | Atypical (single case) | [28] |
| Receptors | ||||
| GABRB2 | EIEE2 (#617829) | AD | Typical (single case) | [11] |
| GABRG2 | DEE74 (#618396) GEFS+ 3 (#607681) FFS8 (#607681) | AD | Atypical (single case) | [11] |
| GABRB3 | DEE43 (#617113) Susceptibility to CAE, type 5 (#612269) | AD | RTT-like (single case) | [46] |
| GABBR2 | DEE59 (#617904) NDD with poor language and loss of hand skills (#617903) | AD | Atypical (single case) | [26] |
| GRIA2 | NDD with language impairment and behavioral abnormalities (#618917) | AD | RTT-like (single case) | [47] |
| GRIN1 | NDD with/out hyperkinetic movements and seizures, AD (#614254) and AR (#617820) | AD, AR | RTT-like: 3/3 | [10,25] |
| GRIN2B | DEE27 (#616139) MR AD 6 (#613970) | AD | RTT-like: 2/2 | [36,48] |
| Transporters | ||||
| SLC6A1 | Myoclonic-atonic epilepsy (#616421) | AD | RTT-like: 1/2 Atypical: 1/2 | [26,36] |
| SLC35A2 | CDG type II (#300896) | XLD, somatic mosaicism | RTT-like (single case) | [9] |
| Transcription Factors/Chromatin Modulation Pathways | ||||
| MEF2C | MR, stereotypic movements, epilepsy and/or cerebral malformations (#613443) | AD | RTT-like: 8/9 RTT: 1/9 | [42,49,50,51] |
| ACTL6B | DEE76 (#618468) Intellectual developmental disorder with severe speech and ambulation defects (#618470) | ARAD | RTT-like (single case) | [52] |
| HDAC4 | CdLS (#300882) | AD de novo deletion chr2.q37.1-q37.3 (including HDAC4)S | Atypical (single case) | [26] |
| HDAC8 | CdLS (#300882) | XLD | RTT-like (single case) | [33] |
| MEIS2 | Cleft palate, cardiac defects, and MR (#600987) | AD | RTT-like (single case) | [41] |
| Ubiquitination | ||||
| RHOBTB2 | DEE64 (#618004) | AD | RTT-like (single case) | [9] |
| HECW2 | NDD with hypotonia, seizures and absent language (#617268) | AD | RTT-like (single case) | [53] |
| Axon Guidance | ||||
| NTNG1 | No OMIM disorder (gene number *608818) | AD | Atypical: 1/2 RTT-like: 1/2 | [26,54] |
| ID + E Genes | ||||
| IQSEC2 | MR, X-linked 1/78 (#309530) | XLD | RTT-like: 21/21 | [55,56,57,58,59] |
| HNRNPH2 | MR, X-linked, syndromic, Bain type (#300986) | XLD | RTT-like (single case) | [58] |
| EEF1A2 | DEE33 (#616409) MR, AD 38 (#616393) | AD AD | RTT-like (single case) | [9] |
| JMJD1C | No OMIM disorder (gene number *604503) | AD | Typical (single case) | [12] |
| To Be Further Evaluated | ||||
| ANXA11 | ALS 23 (#617839) | AD | Atypical (single case) | [26] |
| KIF4B | No OMIM disorder (gene number *609184) | AD | Atypical (single case) | [26] |
| RRN3 | No OMIM disorder (gene number *605121) | AD | Atypical (single case) | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spagnoli, C.; Fusco, C.; Pisani, F. Rett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review. Genes 2021, 12, 1157. https://doi.org/10.3390/genes12081157
Spagnoli C, Fusco C, Pisani F. Rett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review. Genes. 2021; 12(8):1157. https://doi.org/10.3390/genes12081157
Chicago/Turabian StyleSpagnoli, Carlotta, Carlo Fusco, and Francesco Pisani. 2021. "Rett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review" Genes 12, no. 8: 1157. https://doi.org/10.3390/genes12081157
APA StyleSpagnoli, C., Fusco, C., & Pisani, F. (2021). Rett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review. Genes, 12(8), 1157. https://doi.org/10.3390/genes12081157