
Post-Translational Modification of MRE11: Its Implication in DDR and Diseases

 , , and
, , and
Abstract
:1. Introduction
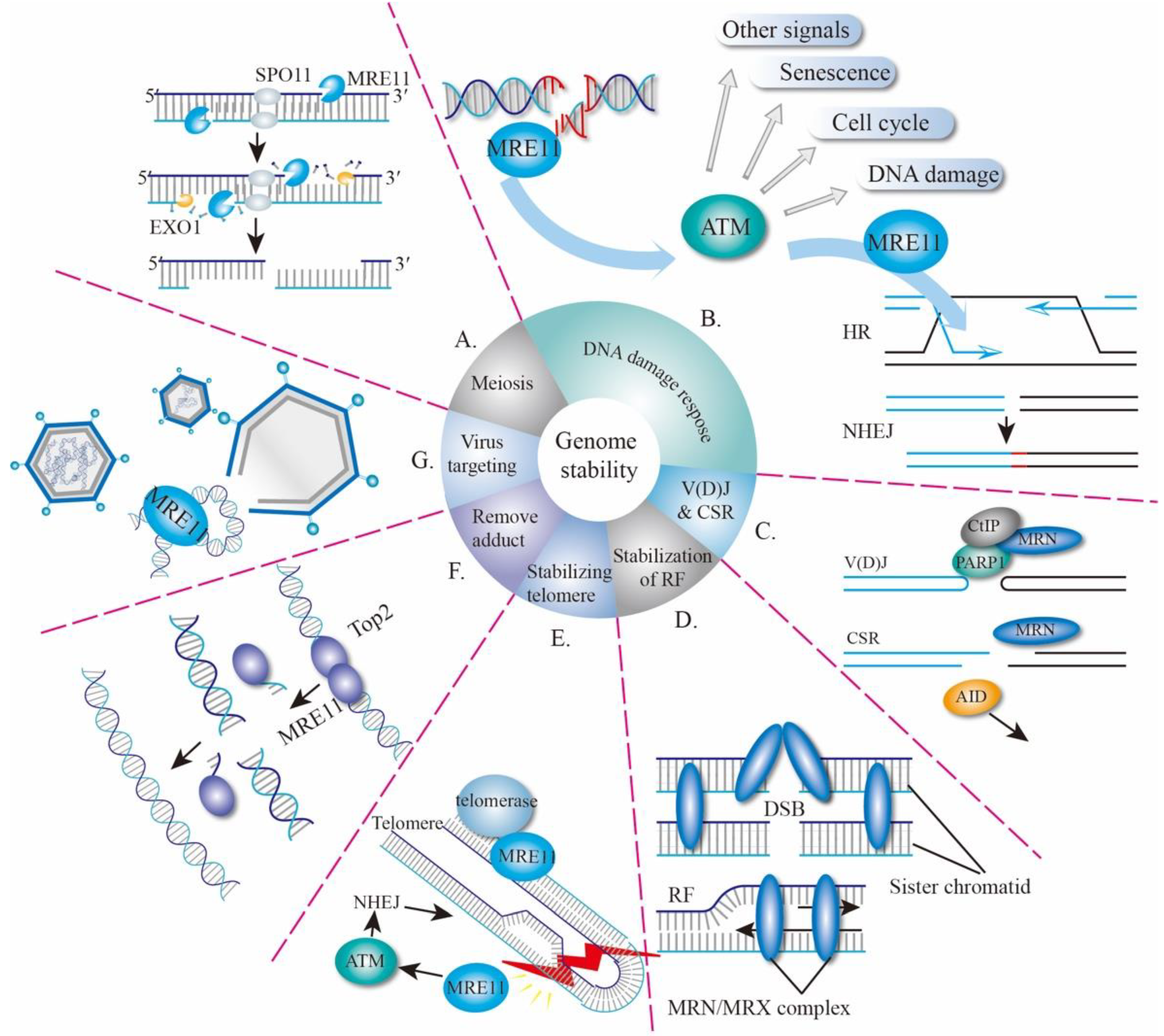
2. Biological Functions of MRE11
2.1. MRE11 in Meiosis
2.2. MRE11 and DNA Damage Response
2.3. MRE11 in V(D)J Recombination and CSR
2.4. Other Function of MRE11
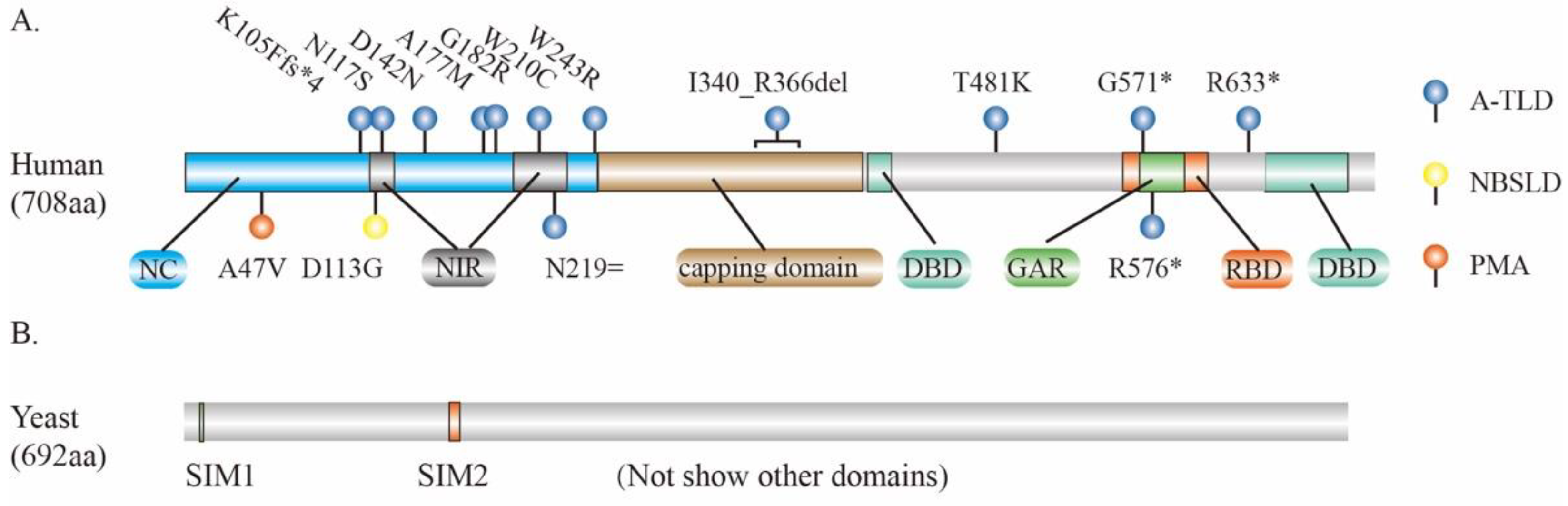
3. MRE11 and Human Diseases
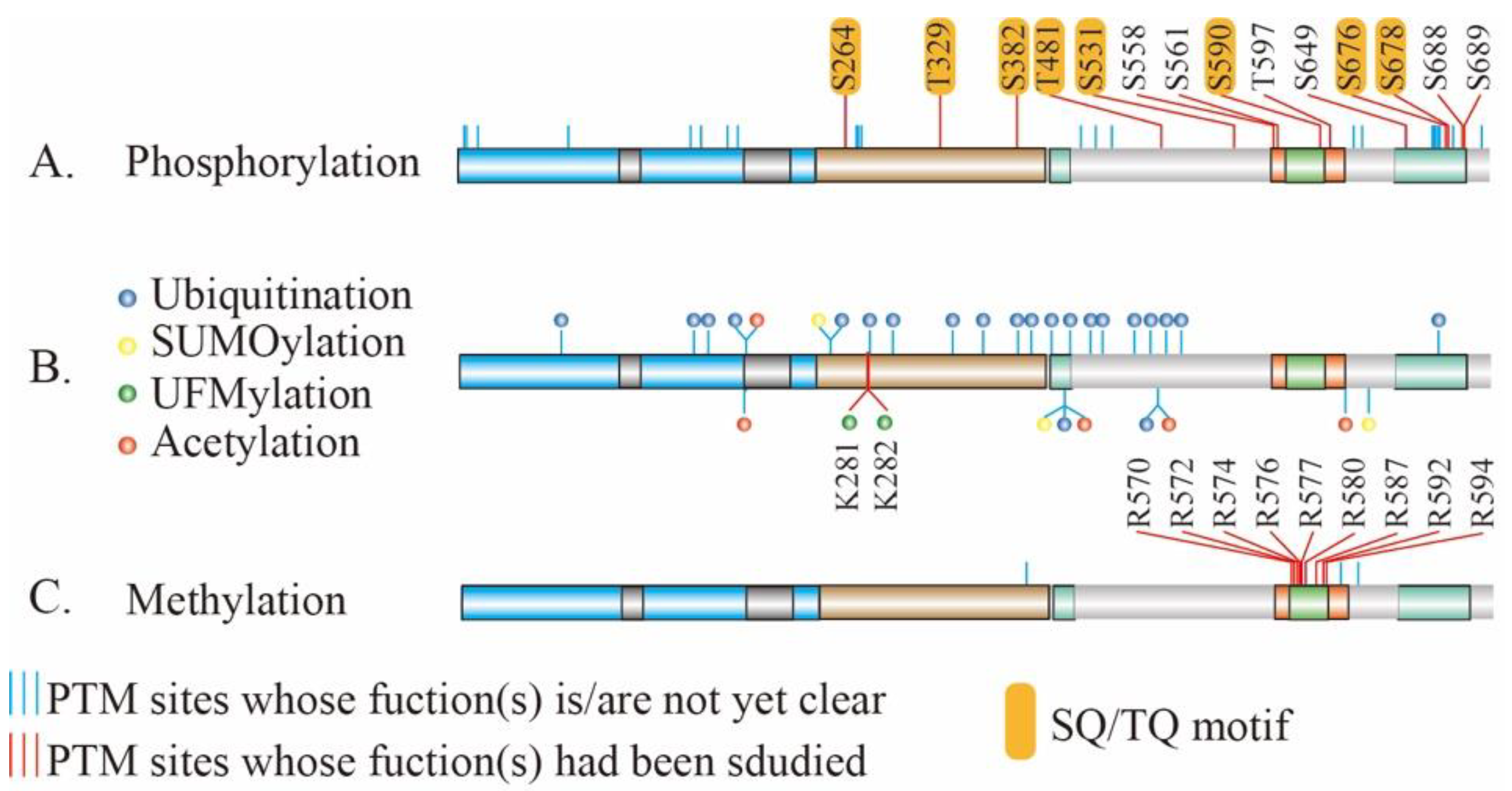
4. PTMs of MRE11 and Their Biological and Pathological Function
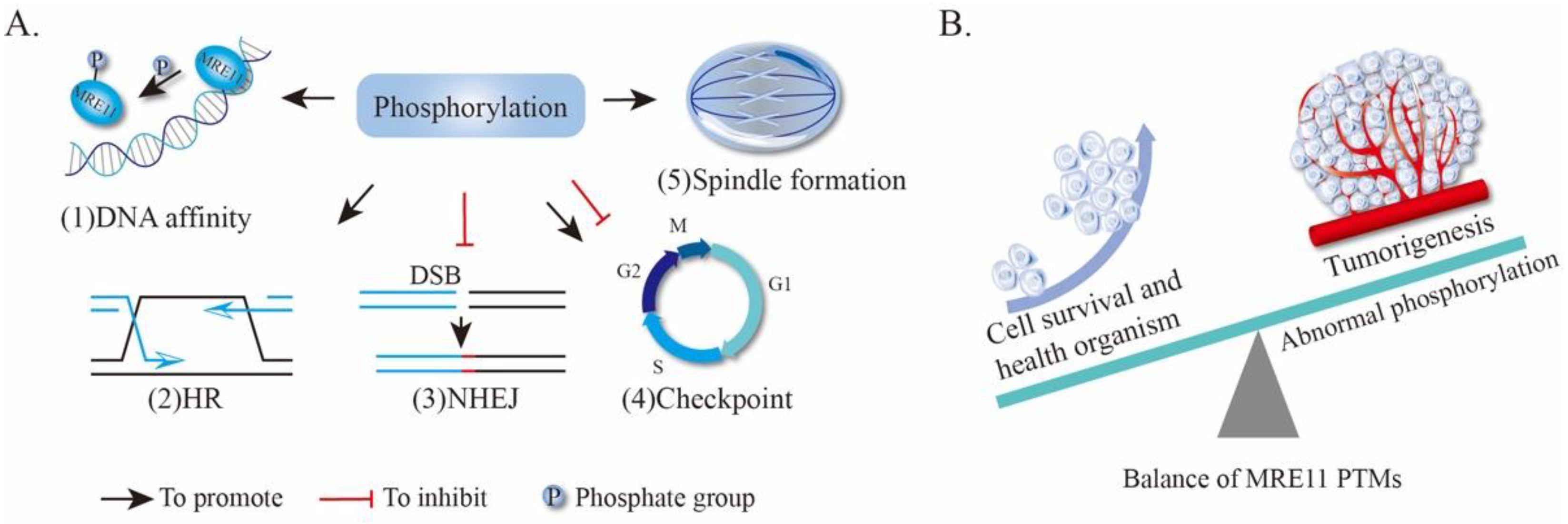
4.1. Phosphorylation of MRE11
4.1.1. MRE11 Phosphorylation Alters the DNA-Binding Capacity of MRE11
4.1.2. Phosphorylation of MRE11 Regulates HR and NHEJ Repair Pathway Choice
4.1.3. MRE11 Phosphorylation Affects Cell Cycle and Chromosomal Alignment
4.1.4. MRE11 Phosphorylation in Tumorigenesis
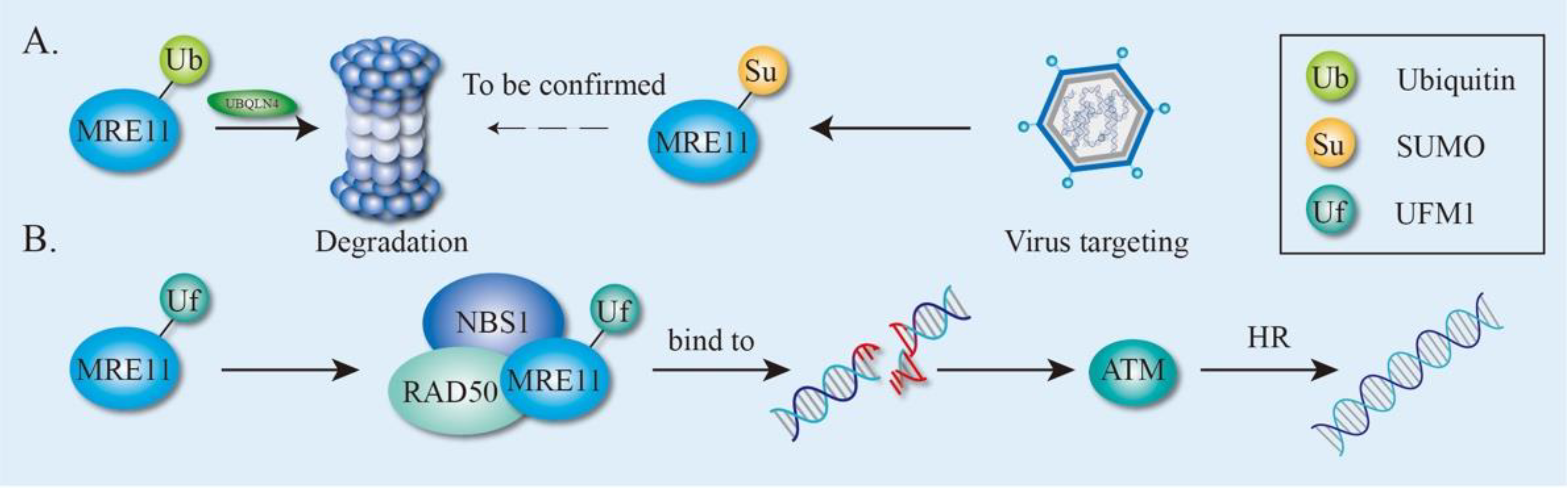
4.2. Ubiquitination and Ubiquitin-Like modification of MRE11
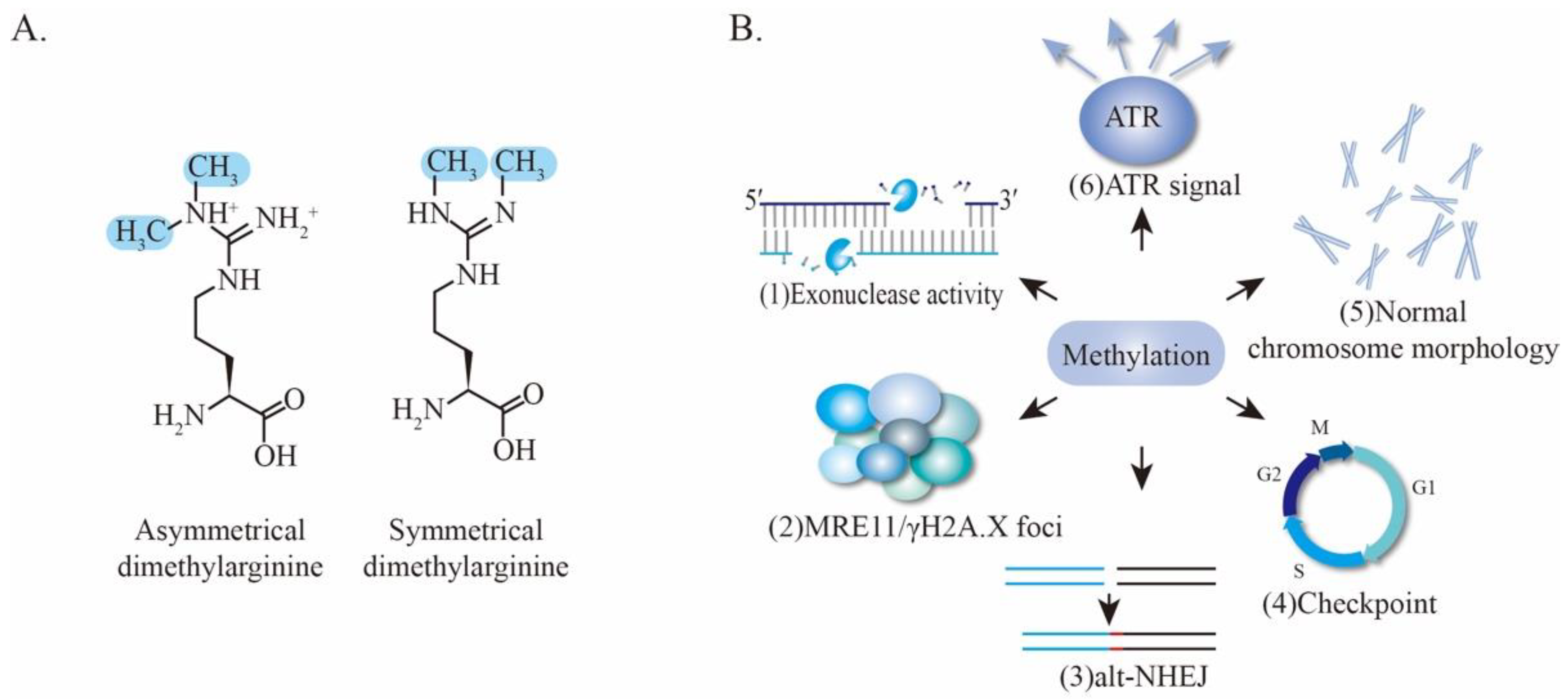
4.3. Methylation of MRE11
5. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, J.F. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res. USA Mol. Biol. 1988, 35, 95–125. [Google Scholar]
- Dedon, P.C. The Chemical Toxicology of 2-Deoxyribose Oxidation in DNA. Chem. Res. Toxicol. 2008, 21, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, G.J.; Hernon, C.; Austriaco, N.; Shapiro, R.S.; Belenky, P.; Bennett, R.J. Metabolism-induced oxidative stress and DNA damage selectively trigger genome instability in polyploid fungal cells. EMBO J. 2019, 38, e101597. [Google Scholar] [CrossRef]
- Engin, A.B.; Engin, A. DNA damage checkpoint response to aflatoxin B1. Environ. Toxicol. Pharm. 2019, 65, 90–96. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, K.; English, N.; Meers, C.; Kim, H.; Jonke, A.; Storici, F.; Torres, M. Systematic analysis of linker histone PTM hotspots reveals phosphorylation sites that modulate homologous recombination and DSB repair. DNA Repair 2020, 86, 102763. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end-joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [Green Version]
- Bruhn, C.; Zhou, Z.W.; Ai, H.; Wang, Z.Q. The essential function of the MRN complex in the resolution of endogenous replication intermediates. Cell Rep. 2014, 6, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Hartlerode, A.J.; Regal, J.A.; Ferguson, D.O. Reversible mislocalization of a disease-associated MRE11 splice variant product. Sci. Rep. 2018, 8, 10121. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska, K.H.; Gregorek, H.; Dembowska-Baginska, B.; Kalina, M.A.; Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 2012, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Canny, M.D.; Buschmann, T.A.; Latham, M.P. A Survey of Reported Disease-Related Mutations in the MRE11-RAD50-NBS1 Complex. Cells 2020, 9, 1678. [Google Scholar] [CrossRef]
- Situ, Y.; Chung, L.; Lee, C.S.; Ho, V. MRN (MRE11-RAD50-NBS1) Complex in Human Cancer and Prognostic Implications in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 816. [Google Scholar] [CrossRef] [Green Version]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol 2021, 11. [Google Scholar] [CrossRef]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, H.; Mair, W.; Kumar, M.; Schlaffner, C.N.; Tang, S.; Beerepoot, P.; Fatou, B.; Guise, A.J.; Cheng, L.; Takeda, S.; et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 2020, 183, 1699–1713. [Google Scholar] [CrossRef]
- Ajimura, M.; Leem, S.H.; Ogawa, H. Identification of New Genes Required for Meiotic Recombination in Saccharomyces cerevisiae. Genetics 1993, 133, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Karcher, A.; Shin, D.; Fairley, C.; Tainer, J.A.; Carney, J.P. Mre11 and Rad50 from Pyrococcus furiosus: Cloning and biochemical characterization reveal an evolutionarily conserved multiprotein machine. J. Bacteriol. 2000, 182, 6036–6041. [Google Scholar] [CrossRef] [Green Version]
- Sharples, G.J.; Leach, D.R. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol. Microbiol. 1995, 17, 1215–1217. [Google Scholar] [CrossRef]
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-Specific DNA Double-Strand Breaks Are Catalyzed by Spo11, a Member of a Widely Conserved Protein Family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Garcia, V.; Phelps, S.E.L.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimitou, E.P.; Yamada, S.; Keeney, S. A global view of meiotic double-strand break end resection. Science 2017, 355, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Petsalaki, E.; Zachos, G. DNA damage response proteins regulating mitotic cell division: Double agents preserving genome stability. FEBS J. 2020, 287, 1700–1721. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.; Kozlov, S.; Gatei, M.; Kijas, A. ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules 2015, 5, 2877–2902. [Google Scholar] [CrossRef] [Green Version]
- Cassani, C.; Vertemara, J.; Bassani, M.; Marsella, A.; Tisi, R.; Zampella, G.; Longhese, M.P. The ATP-bound conformation of the Mre11–Rad50 complex is essential for Tel1/ATM activation. Nucleic Acids Res. 2019, 47, 3550–3567. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gong, Y.; Peng, B.; Shi, R.; Fan, D.; Zhao, H.; Zhu, M.; Zhang, H.; Lou, Z.; Zhou, J.; et al. MRE11 UFMylation promotes ATM activation. Nucleic Acids Res. 2019, 47, 4124–4135. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, J.; Kong, Y.; Yan, S.; Ahmad, N.; Liu, X. Plk1 Phosphorylation of Mre11 Antagonizes the DNA Damage Response. Cancer Res. 2017, 77, 3169–3180. [Google Scholar] [CrossRef] [Green Version]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Kwok, A.; Scully, R. Role of mammalian Mre11 in classical and alternative nonhomologous end-joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef] [Green Version]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.H.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End-joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.; Li, Y.; Qiu, X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: Mechanism and regulation. Immunology 2020, 160, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, D.D.; Chaudhuri, J.; Bassing, C.H.; Alt, F.W. Mechanism and control of V(D)J recombination versus class switch recombination: Similarities and differences. Adv. Immunol. 2005, 86, 43–112. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Bredemeyer, A.L.; Lee, B.S.; Huang, C.Y.; Sharma, G.G.; Walker, L.M.; Bednarski, J.J.; Lee, W.L.; Pandita, T.K.; Bassing, C.H.; et al. MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. J. Exp. Med. 2009, 206, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Boboila, C.; Alt, F.W.; Schwer, B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012, 116, 1–49. [Google Scholar] [CrossRef]
- Seeber, A.; Hegnauer, A.M.; Hustedt, N.; Deshpande, I.; Poli, J.; Eglinger, J.; Pasero, P.; Gut, H.; Shinohara, M.; Hopfner, K.; et al. RPA Mediates Recruitment of MRX to Forks and Double-Strand Breaks to Hold Sister Chromatids Together. Mol. Cell 2016, 64, 951–966. [Google Scholar] [CrossRef] [Green Version]
- Marini, F.; Rawal, C.C.; Liberi, G.; Pellicioli, A. Regulation of DNA Double Strand Breaks Processing: Focus on Barriers. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Trenz, K.; Costanzo, V.; Smith, S. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006, 25, 1764–1774. [Google Scholar] [CrossRef] [Green Version]
- Rozier, L.; Guo, Y.; Peterson, S.; Sato, M.; Baer, R.; Gautier, J.; Mao, Y. The MRN-CtIP pathway is required for metaphase chromosome alignment. Mol. Cell 2013, 49, 1097–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Xu, Y.; Huo, W.; Lv, Z.; Yuan, J.; Ning, S.; Wang, Q.; Hou, M.; Gao, G.; Ji, J.; et al. Mitosis-specific MRN complex promotes a mitotic signaling cascade to regulate spindle dynamics and chromosome segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E10079–E10088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwooll, C.L.; Akpinar, M.; Petrini, J.H.J. The Mre11 Complex and the Response to Dysfunctional Telomeres. Mol. Cell Biol. 2009, 29, 5540–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Liu, J.; Zhang, Q.; Lv, D.; Wu, N.; Zhou, J. Rad6-Bre1-mediated H2B ubiquitination regulates telomere replication by promoting telomere-end resection. Nucleic Acids Res. 2017, 45, 3308–3322. [Google Scholar] [CrossRef] [Green Version]
- Wellinger, R.J.; Zakian, V.A. Everything You Ever Wanted to Know AboutSaccharomyces cerevisiae Telomeres: Beginning to End. Genetics 2012, 191, 1073–1105. [Google Scholar] [CrossRef] [Green Version]
- Lengsfeld, B.M.; Rattray, A.J.; Bhaskara, V.; Ghirlando, R.; Paull, T.T. Sae2 Is an Endonuclease that Processes Hairpin DNA Cooperatively with the Mre11/Rad50/Xrs2 Complex. Mol. Cell 2007, 28, 638–651. [Google Scholar] [CrossRef] [Green Version]
- Lobachev, K.S.; Gordenin, D.A.; Resnick, M.A. The Mre11 Complex Is Required for Repair of Hairpin-Capped Double-Strand Breaks and Prevention of Chromosome Rearrangements. Cell 2002, 108, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, T.; Baer, R.; Gottesman, M.; Gautier, J. MRN, CtIP, and BRCA1 mediate repair of topoisomerase II–DNA adducts. J. Cell Biol. 2016, 212, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoa, N.N.; Shimizu, T.; Zhou, Z.W.; Wang, Z.; Deshpande, R.A.; Paull, T.T.; Akter, S.; Tsuda, M.; Furuta, R.; Tsutsui, K.; et al. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell 2016, 64, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Shah, G.A.; O’Shea, C.C. Viral and Cellular Genomes Activate Distinct DNA Damage Responses. Cell 2015, 162, 987–1002. [Google Scholar] [CrossRef] [Green Version]
- Hopfner, K.P.; Karcher, A.; Craig, L.; Woo, T.T.; Carney, J.P.; Tainer, J.A. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 2001, 105, 473–485. [Google Scholar] [CrossRef] [Green Version]
- Das, D.; Moiani, D.; Axelrod, H.L.; Miller, M.D.; McMullan, D.; Jin, K.K.; Abdubek, P.; Astakhova, T.; Burra, P.; Carlton, D.; et al. Crystal structure of the first eubacterial Mre11 nuclease reveals novel features that may discriminate substrates during DNA repair. J. Mol. Biol. 2010, 397, 647–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.B.; Chae, J.; Kim, Y.C.; Cho, Y. Crystal structure of human Mre11: Understanding tumorigenic mutations. Structure 2011, 19, 1591–1602. [Google Scholar] [CrossRef] [Green Version]
- Tisi, R.; Vertemara, J.; Zampella, G.; Longhese, M.P. Functional and structural insights into the MRX/MRN complex, a key player in recognition and repair of DNA double-strand breaks. Comput. Struct. Biotech. 2020, 18, 1137–1152. [Google Scholar] [CrossRef]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 Dimers Coordinate DNA End Bridging and Nuclease Processing in Double-Strand-Break Repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, R.; Morino, H.; Yoshizawa, A.; Miyazaki, Y.; Maruyama, H.; Murakami, N.; Fukada, K.; Izumi, Y.; Matsuura, S.; Kaji, R.; et al. Exome sequencing reveals a novel MRE11 mutation in a patient with progressive myoclonic ataxia. J. Neurol. Sci. 2014, 337, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Miyamoto, T.; Sakamoto, H.; Izumi, H.; Nakazawa, Y.; Ogi, T.; Tahara, H.; Oku, S.; Hiramoto, A.; Shiiki, T. Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA Repair 2011, 10, 314–321. [Google Scholar] [CrossRef]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.J.; Raams, A.; Byrd, P.J.; Petrini, J.H.J.; Taylor, A.M.R. The DNA Double-Strand Break Repair Gene hMRE11 Is Mutated in Individuals with an Ataxia-Telangiectasia-like Disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Fernet, M.; Gribaa, M.; Salih, M.A.; Seidahmed, M.Z.; Hall, J.; Koenig, M. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum. Mol. Genet. 2005, 14, 307–318. [Google Scholar] [CrossRef]
- Uchisaka, N.; Takahashi, N.; Sato, M.; Kikuchi, A.; Mochizuki, S.; Imai, K.; Nonoyama, S.; Ohara, O.; Watanabe, F.; Mizutani, S.; et al. Two Brothers with Ataxia-Telangiectasia-like Disorder with Lung Adenocarcinoma. J. Pediatrics 2009, 155, 435–438. [Google Scholar] [CrossRef]
- Delia, D.; Piane, M.; Buscemi, G.; Savio, C.; Palmeri, S.; Lulli, P.; Carlessi, L.; Fontanella, E.; Chessa, L. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum. Mol. Genet. 2004, 13, 2155–2163. [Google Scholar] [CrossRef]
- Fiévet, A.; Bellanger, D.; Valence, S.; Mobuchon, L.; Afenjar, A.; Giuliano, F.; Dubois D’Enghien, C.; Parfait, B.; Pedespan, J.M.; Auger, N.; et al. Three new cases of ataxia-telangiectasia-like disorder: No impairment of the ATM pathway, but S-phase checkpoint defect. Hum. Mutat. 2019, 40, 1690–1699. [Google Scholar] [CrossRef]
- Sedghi, M.; Salari, M.; Moslemi, A.; Kariminejad, A.; Davis, M.; Goullée, H.; Olsson, B.; Laing, N.; Tajsharghi, H. Ataxia-telangiectasia-like disorder in a family deficient for MRE11A, caused by aMRE11 variant. Neurol. Genet. 2018, 4, e295. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, E.; Beach, T.; Shen, Y.; Li, R.; Chang, Y. Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Brain Res. Mol. Brain Res. 2004, 128, 1–7. [Google Scholar] [CrossRef]
- Yuan, S.S.; Hou, M.F.; Hsieh, Y.C.; Huang, C.Y.; Lee, Y.C.; Chen, Y.J.; Lo, S. Role of MRE11 in cell proliferation, tumor invasion, and DNA repair in breast cancer. J. Natl. Cancer Inst. 2012, 104, 1485–1502. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Su, T.; Yang, L.; Zhang, C.; He, Y. High expression of MRE11 correlates with poor prognosis in gastric carcinoma. Diagn. Pathol. 2019, 14, 60. [Google Scholar] [CrossRef]
- Pavelitz, T.; Renfro, L.; Foster, N.R.; Caracol, A.; Welsch, P.; Lao, V.V.; Grady, W.B.; Niedzwiecki, D.; Saltz, L.B.; Bertagnolli, M.M.; et al. MRE11-deficiency associated with improved long-term disease free survival and overall survival in a subset of stage III colon cancer patients in randomized CALGB 89803 trial. PLoS ONE 2014, 9, e108483. [Google Scholar] [CrossRef]
- Wang, J.; Xu, W.H.; Wei, Y.; Zhu, Y.; Qin, X.J.; Zhang, H.L.; Ye, D.W. Elevated MRE11 expression associated with progression and poor outcome in prostate cancer. J. Cancer 2019, 10, 4333–4340. [Google Scholar] [CrossRef]
- Spehalski, E.; Capper, K.M.; Smith, C.J.; Morgan, M.J.; Dinkelmann, M.; Buis, J.; Sekiguchi, J.M.; Ferguson, D.O. MRE11 Promotes Tumorigenesis by Facilitating Resistance to Oncogene-Induced Replication Stress. Cancer Res. 2017, 77, 5327–5338. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xin, X.; Chen, Q.; Xie, Z.; Gui, M.; Chen, Y.; Lin, L.; Feng, J.; Li, Q.; Ding, J.; et al. Oligomannurarate sulfate sensitizes cancer cells to doxorubicin by inhibiting atypical activation of NF-kappaB via targeting of Mre11. Int. J. Cancer 2012, 130, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Petroni, M.; Sardina, F.; Infante, P.; Bartolazzi, A.; Locatelli, E.; Fabretti, F.; Di Giulio, S.; Capalbo, C.; Cardinali, B.; Coppa, A.; et al. MRE11 inhibition highlights a replication stress-dependent vulnerability of MYCN-driven tumors. Cell Death Dis. 2018, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 years of PhosphoSitePlus®: Integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019, 47, D433–D441. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.F.; Su, J.; Hou, M.; Yang, F.; Zhao, S.; Lee, E.Y.H.P. Arsenic-induced Mre11 phosphorylation is cell cycle-dependent and defective in NBS cells. DNA Repair 2002, 1, 137–142. [Google Scholar] [CrossRef]
- Yuan, S.F.; Chang, H.; Hou, M.; Chan, T.; Kao, Y.; Wu, Y.; Su, J. Neocarzinostatin induces Mre11 phosphorylation and focus formation through an ATM- and NBS1-dependent mechanism. Toxicology 2002, 177, 123–130. [Google Scholar] [CrossRef]
- Kijas, A.W.; Lim, Y.C.; Bolderson, E.; Cerosaletti, K.; Gatei, M.; Jakob, B.; Tobias, F.; Taucher-Scholz, G.; Gueven, N.; Oakley, G.; et al. ATM-dependent phosphorylation of MRE11 controls extent of resection during homology directed repair by signalling through Exonuclease 1. Nucleic Acids Res. 2015, 43, 8352–8367. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, M.; Ying, C.Y.; Gautier, J. PIKK-dependent phosphorylation of Mre11 induces MRN complex inactivation by disassembly from chromatin. DNA Repair 2009, 8, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Zhong, Q.; Chen, P.L. The Nijmegen breakage syndrome protein is essential for Mre11 phosphorylation upon DNA damage. J. Biol. Chem. 1999, 274, 19513–19516. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Wang, W.; Li, S.; Zhan, J.; Li, H.; Zhao, M.; Zhou, X.A.; Li, S.; Li, X.; Huo, Y.; et al. C1QBP Promotes Homologous Recombination by Stabilizing MRE11 and Controlling the Assembly and Activation of MRE11/RAD50/NBS1 Complex. Mol. Cell 2019, 75, 1299–1314. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Jachimowicz, R.D.; Beleggia, F.; Isensee, J.; Velpula, B.B.; Goergens, J.; Bustos, M.A.; Doll, M.A.; Shenoy, A.; Checa-Rodriguez, C.; Wiederstein, J.L.; et al. UBQLN4 Represses Homologous Recombination and Is Overexpressed in Aggressive Tumors. Cell 2019, 176, 505–519. [Google Scholar] [CrossRef] [Green Version]
- Simoneau, A.; Robellet, X.; Ladouceur, A.M.; D’Amours, D. Cdk1-dependent regulation of the Mre11 complex couples DNA repair pathways to cell cycle progression. Cell Cycle 2014, 13, 1078–1090. [Google Scholar] [CrossRef] [Green Version]
- Kim, S. Protein kinase CK2 interacts with Chk2 and phosphorylates Mre11 on serine 649. Biochem. Biophys. Res. Commun. 2005, 331, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, L.; Huang, N.J.; Huang, B.; Kornbluth, S. Suppression of DNA-damage checkpoint signaling by Rsk-mediated phosphorylation of Mre11. Proc. Natl. Acad. Sci. USA 2013, 110, 20605–20610. [Google Scholar] [CrossRef] [Green Version]
- Piscitello, D.; Varshney, D.; Lilla, S.; Vizioli, M.G.; Reid, C.; Gorbunova, V.; Seluanov, A.; Gillespie, D.A.; Adams, P.D. AKT overactivation can suppress DNA repair via p70S6 kinase-dependent downregulation of MRE11. Oncogene 2018, 37, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Li, S. The Role of Ubiquitination in NF-kappaB Signaling during Virus Infection. Viruses 2021, 13, 145. [Google Scholar] [CrossRef]
- Su, S.; Zhang, Y.; Liu, P. Roles of Ubiquitination and SUMOylation in DNA Damage Response. Curr. Issues. Mol. Biol. 2020, 35, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Shoji, Y.; Nishi, T.; Chang, S.C.; Jachimowicz, R.D.; Hoshimoto, S.; Ono, S.; Shiloh, Y.; Takeuchi, H.; Kitagawa, Y.; et al. Regulation of MRE11A by UBQLN4 leads to cisplatin-resistance in patients with esophageal squamous cell carcinoma. Mol. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.; Jevons, S.J.; Groselj, B.; Ellermann, S.; Konietzny, R.; Kerr, M.; Kessler, B.M.; Kiltie, A.E. E3 Ligase cIAP2 Mediates Downregulation of MRE11 and Radiosensitization in Response to HDAC Inhibition in Bladder Cancer. Cancer Res. 2017, 77, 3027–3039. [Google Scholar] [CrossRef] [Green Version]
- Henley, J.M.; Seager, R.; Nakamura, Y.; Talandyte, K.; Nair, J.; Wilkinson, K.A. SUMOylation of synaptic and synapse-associated proteins: An update. J. Neurochem. 2021, 156, 145–161. [Google Scholar] [CrossRef]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chuang, Y.; Chuang, C.; Cheng, Y.; Chang, C.; Leng, C.; Wang, T.S. cerevisiae Mre11 recruits conjugated SUMO moieties to facilitate the assembly and function of the Mre11-Rad50-Xrs2 complex. Nucleic Acids Res. 2016, 44, 2199–2213. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Goben, B.; Meisenhelder, J.; You, Z.; Weitzman, M.D.; Hunter, T. Repair of protein-linked DNA double strand breaks: Using the adenovirus genome as a model substrate in cell-based assays. DNA Repair 2019, 74, 80–90. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Ornelles, D.A. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene 2005, 24, 7686–7696. [Google Scholar] [CrossRef] [Green Version]
- Kleinberger, T. En Guard! The Interactions between Adenoviruses and the DNA Damage Response. Viruses 2020, 12, 996. [Google Scholar] [CrossRef]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef]
- Harada, J.N.; Shevchenko, A.; Shevchenko, A.; Pallas, D.C.; Berk, A.J. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 2002, 76, 9194–9206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, S.; Hearing, P. Adenovirus Regulates Sumoylation of Mre11-Rad50-Nbs1 Components through a Paralog-Specific Mechanism. J. Virol. 2012, 86, 9656–9665. [Google Scholar] [CrossRef] [Green Version]
- Sohn, S.; Hearing, P. Mechanism of Adenovirus E4-ORF3-Mediated SUMO Modifications. mBio 2019, 10, e19–e22. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Kumar, M.; Wiener, R. Decrypting UFMylation: How Proteins Are Modified with UFM1. Biomolecules 2020, 10, 1442. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, W.G.; Xu, X. Ubiquitin-like modifications in the DNA damage response. Mutat. Res. 2017, 803–805, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xu, X. UFMylation: A Unique & Fashionable Modification for Life. Genom. Proteom. Bioinform. 2016, 14, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Perez Oliva, A.B.; Churikov, D.; Martinez-Balsalobre, E.; Peter, J.; Rahmouni, D.; Audoly, G.; Azzoni, V.; Audebert, S.; Camoin, L.; et al. UFMylation of MRE11 is essential for maintenance of telomere length and hematopoietic stem cell survival. BioRxiv 2019, 846477. [Google Scholar] [CrossRef] [Green Version]
- Dilworth, D.; Barsyte-Lovejoy, D. Targeting protein methylation: From chemical tools to precision medicines. Cell Mol. Life Sci. 2019, 76, 2967–2985. [Google Scholar] [CrossRef]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef] [Green Version]
- Maeda, M.; Hasegawa, H.; Sugiyama, M.; Hyodo, T.; Ito, S.; Chen, D.; Asano, E.; Masuda, A.; Hasegawa, Y.; Hamaguchi, M.; et al. Arginine methylation of ubiquitin-associated protein 2-like is required for the accurate distribution of chromosomes. FASEB J. 2016, 30, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Bedford, M.T.; Richard, S. Arginine methylation an emerging regulator of protein function. Mol. Cell 2005, 18, 263–272. [Google Scholar] [CrossRef]
- Boisvert, F.; Côté, J.; Boulanger, M.; Richard, S. A Proteomic Analysis of Arginine-methylated Protein Complexes. Mol. Cell Proteom. 2003, 2, 1319–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisvert, F.M.; Dery, U.; Masson, J.Y.; Richard, S. Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes Dev. 2005, 19, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, F.; Hendzel, M.J.; Masson, J.; Richard, S. Methylation of MRE11 Regulates its Nuclear Compartmentalization. Cell Cycle 2005, 4, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Kopp, B.; Khoury, L.; Audebert, M. Validation of the gammaH2AX biomarker for genotoxicity assessment: A review. Arch. Toxicol. 2019, 93, 2103–2114. [Google Scholar] [CrossRef]
- Dery, U.; Coulombe, Y.; Rodrigue, A.; Stasiak, A.; Richard, S.; Masson, J.Y. A Glycine-Arginine Domain in Control of the Human MRE11 DNA Repair Protein. Mol. Cell Biol. 2008, 28, 3058–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, A.N.; Bedford, M.T. Protein methylation and DNA repair. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2007, 618, 91–101. [Google Scholar] [CrossRef]
- Choi, K.; Kim, J.Y.; Lim, S.; Choi, Y.W.; Kim, Y.H.; Kang, S.Y.; Park, T.J.; Lim, I.K. TIS21/BTG2/PC3 accelerates the repair of DNA double strand breaks by enhancing Mre11 methylation and blocking damage signal transfer to the Chk2T68–p53S20 pathway. DNA Repair 2012, 11, 965–975. [Google Scholar] [CrossRef]
- Vadnais, C.; Chen, R.; Fraszczak, J.; Yu, Z.; Boulais, J.; Pinder, J.; Frank, D.; Khandanpour, C.; Hébert, J.; Dellaire, G.; et al. GFI1 facilitates efficient DNA repair by regulating PRMT1 dependent methylation of MRE11 and 53BP1. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Zhuang, J.; Jiang, G.; Willers, H.; Xia, F. Exonuclease Function of Human Mre11 Promotes Deletional Nonhomologous End-joining. J. Biol. Chem. 2009, 284, 30565–30573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhenbao, Y.G.V.Y. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res. 2012, 22, 305–320. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Tian, R.; Zhao, H.; Li, L.; Bi, X. Multiple Arginine Residues Are Methylated in Drosophila Mre11 and Required for Survival Following Ionizing Radiation. G3 2018, 8, 2099–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kish, A.; Gaillard, J.; Armengaud, J.; Elie, C. Post-translational methylations of the archaeal Mre11:Rad50 complex throughout the DNA damage response. Mol. Microbiol. 2016, 100, 362–378. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Bu, C.; Liu, Y.; Gong, T.; Liu, X.; Liu, S.; Peng, X.; Zhang, W.; Peng, Y.; Yang, J.; et al. Global crotonylome reveals CDYL-regulated RPA1 crotonylation in homologous recombination-mediated DNA repair. Sci. Adv. 2020, 6, y4697. [Google Scholar] [CrossRef] [Green Version]
- Rechkunova, N.I.; Maltseva, E.A.; Lavrik, O.I. Post-translational Modifications of Nucleotide Excision Repair Proteins and Their Role in the DNA Repair. Biochem. 2019, 84, 1008–1020. [Google Scholar] [CrossRef]
- Ghosh, D.; Bohr, V.A.; Karmakar, P. Acetylation of Werner protein at K1127 and K1117 is important for nuclear trafficking and DNA repair. DNA Repair 2019, 79, 22–31. [Google Scholar] [CrossRef]
- Hou, W.H.; Chen, S.H.; Yu, X. Poly-ADP ribosylation in DNA damage response and cancer therapy. Mutat. Res. 2019, 780, 82–91. [Google Scholar] [CrossRef]
- Baranes-Bachar, K.; Levy-Barda, A.; Oehler, J.; Reid, D.A.; Soria-Bretones, I.; Voss, T.C.; Chung, D.; Park, Y.; Liu, C.; Yoon, J.B.; et al. The Ubiquitin E3/E4 Ligase UBE4A Adjusts Protein Ubiquitylation and Accumulation at Sites of DNA Damage, Facilitating Double-Strand Break Repair. Mol. Cell 2018, 69, 866–878. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef]
- Tammsalu, T.; Matic, I.; Jaffray, E.G.; Ibrahim, A.; Tatham, M.H.; Hay, R.T. Proteome-wide identification of SUMO2 modification sites. Sci. Signal. 2014, 7, s2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupre, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Number of Somatic Mutations * | Affected Cases |
|---|---|---|
| MRE11 | 125 | 840 |
| RAD50 | 229 | 869 |
| NBS1 | 136 | 978 |
| Cancer Type | Protein Change | Mutation Type | Variant Type |
|---|---|---|---|
| Acute Myeloid Leukemia | Q477E | Missense_Mutation | SNP |
| Adrenocortical Carcinoma | R351C | Missense_Mutation | SNP |
| Bladder Urothelial Carcinoma | Q438E | Missense_Mutation | SNP |
| Breast Invasive Ductal Carcinoma | R503C | Missense_Mutation | SNP |
| Cervical Squamous Cell Carcinoma | S641* | Nonsense_Mutation | SNP |
| Cutaneous Melanoma | N511Ifs*13 | Frame_Shift_Del | DEL |
| Glioblastoma Multiforme | R364* | Nonsense_Mutation | SNP |
| Head and Neck Squamous Cell Carcinoma | S608* | Nonsense_Mutation | SNP |
| Lung Squamous Cell Carcinoma | E350* | Nonsense_Mutation | SNP |
| Mucinous Adenocarcinoma of the Colon and Rectum | E460* | Nonsense_Mutation | SNP |
| Pancreatic Adenocarcinoma | D498N | Missense_Mutation | SNP |
| Papillary Renal Cell Carcinoma | L44V | Missense_Mutation | SNP |
| Papillary Thyroid Cancer | R505I | Missense_Mutation | SNP |
| Renal Clear Cell Carcinoma | T627A | Missense_Mutation | SNP |
| Serous Ovarian Cancer | A526Gfs*16 | Frame_Shift_Ins | INS |
| Stomach Adenocarcinoma | N511Ifs*13 | Frame_Shift_Del | DEL |
| Uterine Endometrioid Carcinoma | E460* | Nonsense_Mutation | SNP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, R.; Zhang, H.; Jiang, Y.-N.; Wang, Z.-Q.; Sun, L.; Zhou, Z.-W. Post-Translational Modification of MRE11: Its Implication in DDR and Diseases. Genes 2021, 12, 1158. https://doi.org/10.3390/genes12081158
Lu R, Zhang H, Jiang Y-N, Wang Z-Q, Sun L, Zhou Z-W. Post-Translational Modification of MRE11: Its Implication in DDR and Diseases. Genes. 2021; 12(8):1158. https://doi.org/10.3390/genes12081158
Chicago/Turabian StyleLu, Ruiqing, Han Zhang, Yi-Nan Jiang, Zhao-Qi Wang, Litao Sun, and Zhong-Wei Zhou. 2021. "Post-Translational Modification of MRE11: Its Implication in DDR and Diseases" Genes 12, no. 8: 1158. https://doi.org/10.3390/genes12081158
APA StyleLu, R., Zhang, H., Jiang, Y. -N., Wang, Z. -Q., Sun, L., & Zhou, Z. -W. (2021). Post-Translational Modification of MRE11: Its Implication in DDR and Diseases. Genes, 12(8), 1158. https://doi.org/10.3390/genes12081158