
TWEAKing the Hippocampus: The Effects of TWEAK on the Genomic Fabric of the Hippocampus in a Neuropsychiatric Lupus Mouse Model

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Transcriptomics
3. Results
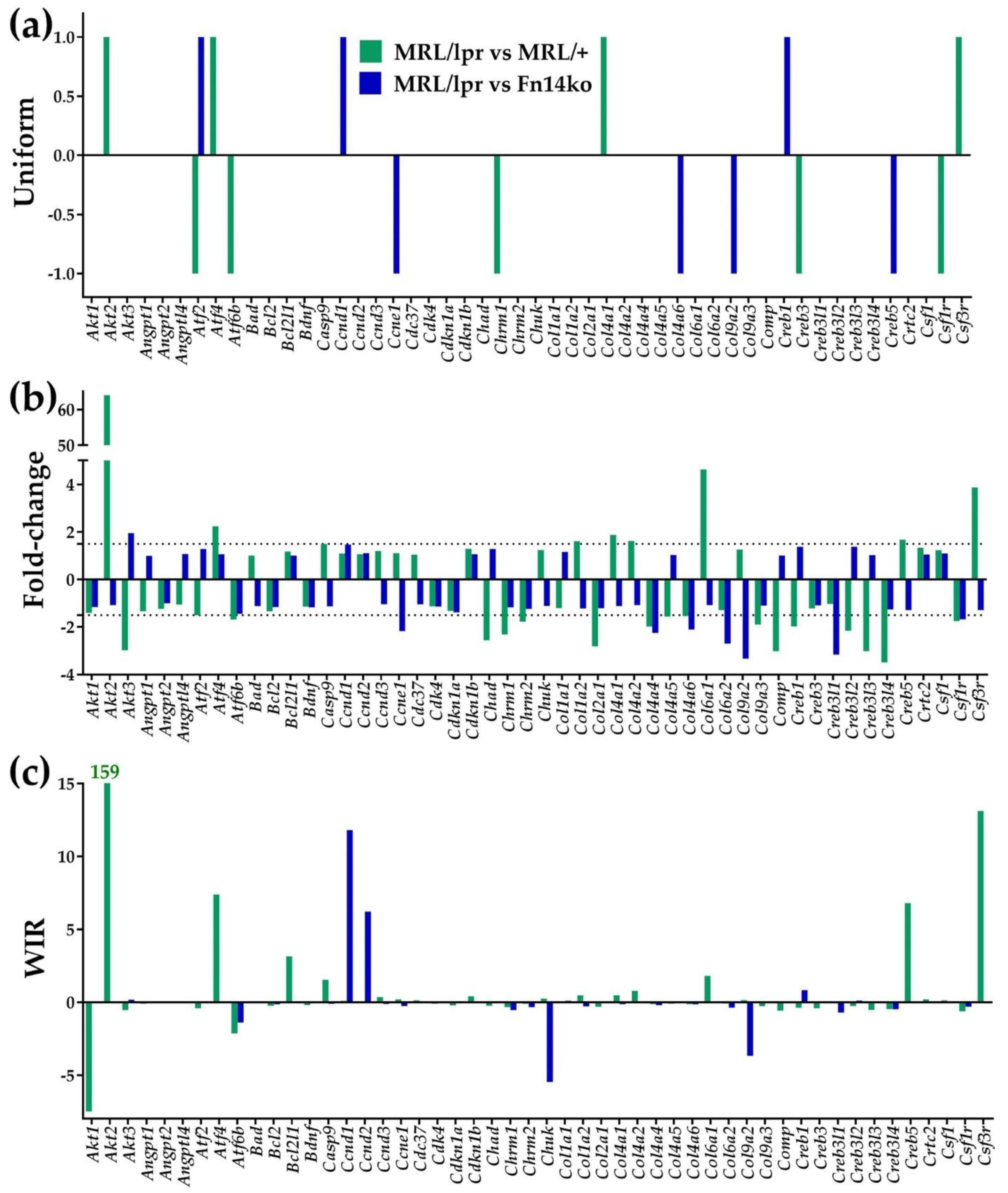
3.1. Independent Expression Characteristics of Individual Genes
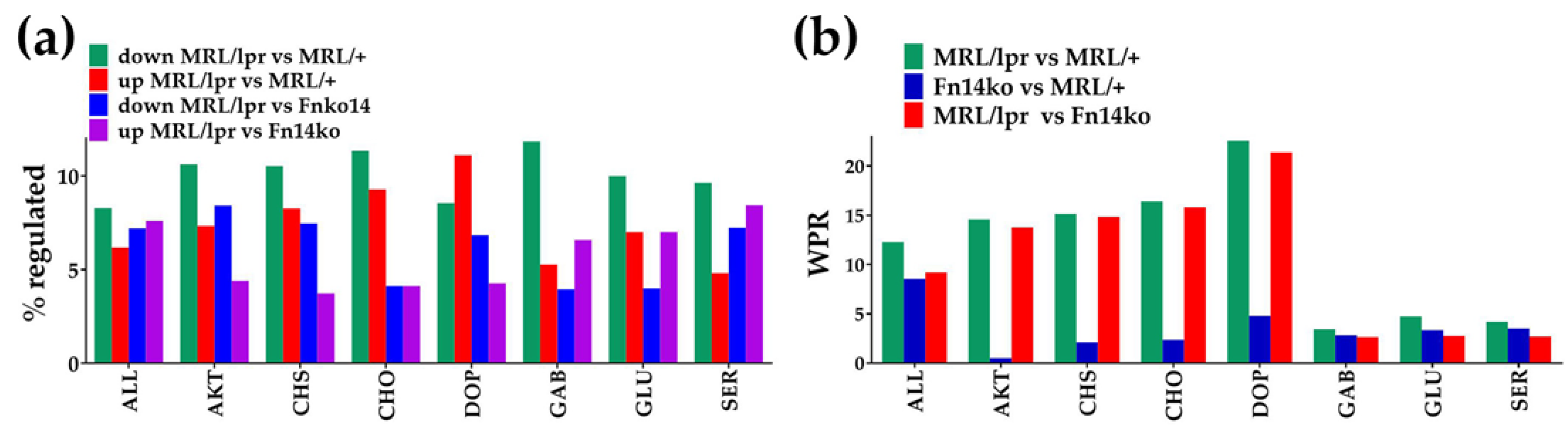
3.2. Measures of Expression Regulation
3.3. Regulation of the Genomic Fabrics Responsible for Neurotransmission, Chemokine Signaling and PI3K-AKT Signaling Pathways
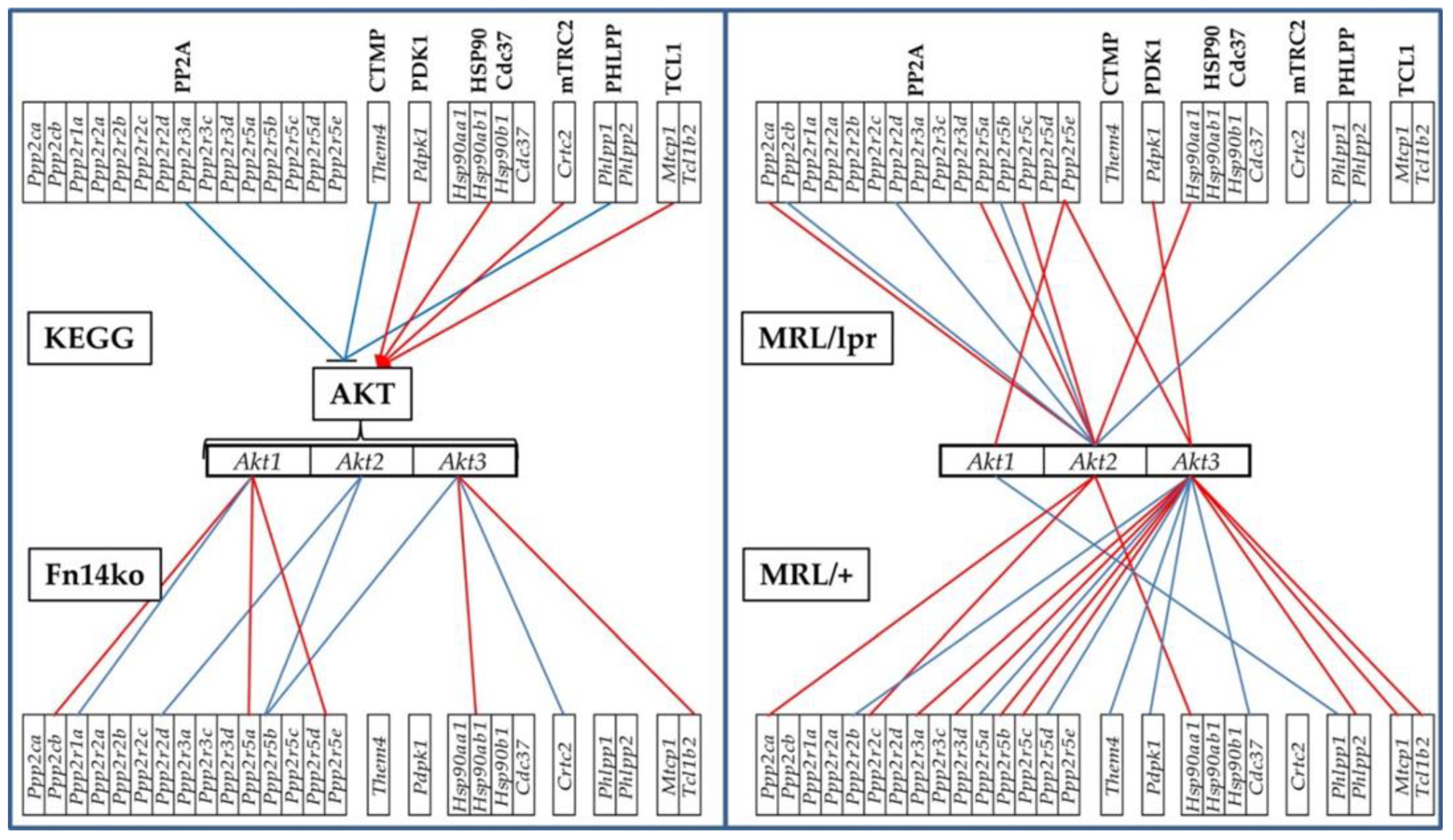
3.4. Gene Hierarchy in the Hippocampus
3.5. Phenotype Dependence of the PI3K-AKT Pathway
3.6. Phenotype-Dependent Cortex-Hippocampus Synchronous Expression of Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schwartz, N.; Stock, A.D.; Putterman, C. Neuropsychiatric lupus: New mechanistic insights and future treatment directions. Nat. Rev. Rheumatol. 2019, 15, 137–152. [Google Scholar] [CrossRef] [PubMed]
- The American College of Rheumatology. Nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999, 42, 599–608. [Google Scholar] [CrossRef]
- Mackay, M.; Vo, A.; Tang, C.C.; Small, M.; Anderson, E.W.; Ploran, E.J.; Storbeck, J.; Bascetta, B.; Kang, S.; Aranow, C.; et al. Metabolic and microstructural alterations in the SLE brain correlate with cognitive impairment. JCI Insight 2019, 4, e124002. [Google Scholar] [CrossRef]
- Steup-Beekman, G.M.; Zirkzee, E.J.; Cohen, D.; Gahrmann, B.M.; Emmer, B.J.; Steens, S.C.; Bollen, E.L.; van Buchem, M.A.; Huizinga, T.W. Neuropsychiatric manifestations in patients with systemic lupus erythematosus: Epidemiology and radiology pointing to an immune-mediated cause. Ann. Rheum. Dis. 2013, 72, ii76–ii79. [Google Scholar] [CrossRef]
- Bertsias, G.K.; Ioannidis, J.P.; Aringer, M.; Bollen, E.; Bombardieri, S.; Bruce, I.N.; Cervera, R.; Dalakas, M.; Doria, A.; Hanly, J.G.; et al. EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: Report of a task force of the EULAR standing committee for clinical affairs. Ann. Rheum. Dis. 2010, 69, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Hanly, J.G.; Kozora, E.; Beyea, S.; Birnbaum, J. Nervous system disease in Systemic Lupus Erythematosus: Current status and future directions. Arthritis Rheumatol. 2018, 71, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kello, N.; Anderson, E.; Diamond, B. Cognitive dysfunction in Systemic Lupus Erythematosus: A case for initiating trials. Arthritis Rheumatol. 2019, 71, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Sanam, S.; Kate, K.; Mohan, C. Animal models of lupus and lupus nephritis. Curr. Pharm. Des. 2015, 21, 2320–2349. [Google Scholar] [CrossRef] [PubMed]
- Ballok, D.A. Neuroimmunopathology in a murine model of neuropsychiatric lupus. Brain Res. Rev. 2007, 54, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulinello, M.; Putterman, C. The MRL/lpr mouse strain as a model for neuropsychiatric Systemic Lupus Erythematosus. J. Biomed. Biotechnol. 2011, 2011, 207504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerner, J.L.; Wen, J.; Xia, Y.; Paz, K.B.; Schairer, D.; Wu, L.; Chalmers, S.A.; Izmirly, P.; Michaelson, J.S.; Burkly, L.C.; et al. TWEAK/Fn14 signaling involvement in the pathogenesis of cutaneous disease in the MRL/lpr model of spontaneous Lupus. J. Investig. Dermatol. 2015, 135, 1986–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelson, J.S.; Wisniacki, N.; Burkly, L.C.; Putterman, C. Role of TWEAK in lupus nephritis: A bench-to-bedside review. J. Autoimmun. 2012, 39, 130–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Burkly, L.C.; Campbell, S.; Schwartz, N.; Molano, A.; Choudhury, A.; Eisenberg, R.A.; Michaelson, J.S.; Putterman, C. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J. Immunol. 2007, 179, 7949–7958. [Google Scholar] [CrossRef] [Green Version]
- Desplat-Jego, S.; Varriale, S.; Creidy, R.; Terra, R.; Bernard, D.; Khrestchatisky, M.; Izui, S.; Chicheportiche, Y.; Boucraut, J. TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. J. Neuroimmunol. 2002, 133, 116–123. [Google Scholar] [CrossRef]
- Stock, A.D.; Wen, J.; Putterman, C. Neuropsychiatric Lupus, the Blood Brain Barrier, and the TWEAK/Fn14 Pathway. Front. Immunol. 2013, 4, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Doerner, J.; Weidenheim, K.; Xia, Y.; Stock, A.; Michaelson, J.S.; Baruch, K.; Deczkowska, A.; Gulinello, M.; Schwartz, M.; et al. TNF-like weak inducer of apoptosis promotes blood brain barrier disruption and increases neuronal cell death in MRL/lpr mice. J. Autoimmun. 2015, 60, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Xia, Y.; Stock, A.; Michaelson, J.S.; Burkly, L.C.; Gulinello, M.; Putterman, C. Neuropsychiatric disease in murine lupus is dependent on the TWEAK/Fn14 pathway. J. Autoimmun. 2013, 43, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Chen, C.H.; Stock, A.; Doerner, J.; Gulinello, M.; Putterman, C. Intracerebroventricular administration of TNF-like weak inducer of apoptosis induces depression-like behavior and cognitive dysfunction in non-autoimmune mice. Brain Behav. Immun. 2016, 54, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Fragoso-Loyo, H.; Cabiedes, J.; Orozco-Narvaez, A.; Davila-Maldonado, L.; Atisha-Fregoso, Y.; Diamond, B.; Llorente, L.; Sanchez-Guerrero, J. Serum and cerebrospinal fluid autoantibodies in patients with neuropsychiatric lupus erythematosus. Implications for diagnosis and pathogenesis. PLoS ONE 2008, 3, e3347. [Google Scholar] [CrossRef] [Green Version]
- Iacobas, D.; Wen, J.; Iacobas, S.; Schwartz, N.; Putterman, C. Remodeling of neurotransmission, chemokine, and PI3K-AKT signaling genomic fabrics in neuropsychiatric Systemic Lupus Erythematosus. Genes 2021, 12, 251. [Google Scholar] [CrossRef]
- Appenzeller, S.; Carnevalle, A.D.; Li, L.M.; Costallat, L.T.; Cendes, F. Hippocampal atrophy in systemic lupus erythematosus. Ann. Rheum. Dis. 2006, 65, 1585–1589. [Google Scholar] [CrossRef] [Green Version]
- Ballok, D.A.; Woulfe, J.; Sur, M.; Cyr, M.; Sakic, B. Hippocampal damage in mouse and human forms of systemic autoimmune disease. Hippocampus 2004, 14, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Kozora, E.; Brown, M.S.; Filley, C.M.; Zhang, L.; Miller, D.E.; West, S.G.; Pelzman, J.; Arciniegas, D.B. Memory impairment associated with neurometabolic abnormalities of the hippocampus in patients with non-neuropsychiatric systemic lupus erythematosus. Lupus 2011, 20, 598–606. [Google Scholar] [CrossRef]
- Lauvsnes, M.B.; Beyer, M.K.; Kvaloy, J.T.; Greve, O.J.; Appenzeller, S.; Kvivik, I.; Harboe, E.; Tjensvoll, A.B.; Goransson, L.G.; Omdal, R. Association of hippocampal atrophy with cerebrospinal fluid antibodies against the NR2 subtype of the N-methyl-D-aspartate receptor in patients with systemic lupus erythematosus and patients with primary Sjogren’s syndrome. Arthritis Rheumatol. 2014, 66, 3387–3394. [Google Scholar] [CrossRef]
- Liu, S.; Cheng, Y.; Zhao, Y.; Lai, A.; Lv, Z.; Xie, Z.; Upreti, B.; Wang, X.; Xu, X.; Luo, C.; et al. Hippocampal atrophy in Systemic Lupus Erythematosus patients without major neuropsychiatric manifestations. J. Immunol. Res. 2020, 2020, 2943848. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, N.; Correa, D.G.; Kubo, T.A.; Netto, T.M.; Pereira, D.B.; Fonseca, R.P.; Gasparetto, E.L. Global cognitive impairment in Systemic Lupus Erythematosus patients: A structural MRI study. Clin. Neuroradiol. 2017, 27, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Iacobas, S.; Stout, R.F.; Spray, D.C. Cellular environment remodels the genomic fabrics of functional pathways in astrocytes. Genes 2020, 11, 520. [Google Scholar] [CrossRef] [PubMed]
- Victorino, P.H.; Marra, C.; Iacobas, D.A.; Iacobas, S.; Spray, D.C.; Linden, R.; Adesse, D.; Petrs-Silva, H. Retinal genomic fabric remodeling after optic nerve injury. Genes 2021, 12, 403. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Lee, P.R.; Cohen, J.E.; Fields, R.D. Coordinated activity of transcriptional networks responding to the pattern of action potential firing in neurons. Genes 2019, 10, 754. [Google Scholar] [CrossRef] [Green Version]
- Law of Multiple Proportions. Available online: https://www.britannica.com/science/law-of-multiple-proportions (accessed on 24 April 2021).
- Iacobas, D.A. Biomarkers, master regulators and genomic fabric remodeling in a case of Papillary Thyroid Carcinoma. Genes 2020, 11, 1030. [Google Scholar] [CrossRef]
- Iacobas, S.; Ede, N.; Iacobas, D.A. The Gene Master Regulators (GMR) approach provides legitimate targets for personalized, time-sensitive cancer gene therapy. Genes 2019, 10, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobas, D.A.; Tuli, N.Y.; Iacobas, S.; Rasamny, J.K.; Moscatello, A.; Geliebter, J.; Tiwari, R.K. Gene master regulators of papillary and anaplastic thyroid cancers. Oncotarget 2018, 9, 2410–2424. [Google Scholar] [CrossRef] [Green Version]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ Proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160534. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Yoshida, S.; Sugeno, N.; Kobayashi, J.; Aoki, M. DnaJ/Hsp40 family and Parkinson’s Disease. Front. Neurosci. 2017, 11, 743. [Google Scholar] [CrossRef]
- Park, S.K.; Arslan, F.; Kanneganti, V.; Barmada, S.J.; Purushothaman, P.; Verma, S.C.; Liebman, S.W. Overexpression of a conserved HSP40 chaperone reduces toxicity of several neurodegenerative disease proteins. Prion 2018, 12, 16–22. [Google Scholar] [CrossRef]
- Tanaka, S.; Kitagawa, K.; Ohtsuki, T.; Yagita, Y.; Takasawa, K.; Hori, M.; Matsumoto, M. Synergistic induction of HSP40 and HSC70 in the mouse hippocampal neurons after cerebral ischemia and ischemic tolerance in gerbil hippocampus. J. Neurosci. Res. 2002, 67, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Guttula, S.V.; Allam, A.; Gumpeny, R.S. Analyzing microarray data of Alzheimer’s using cluster analysis to identify the biomarker genes. Int. J. Alzheimers Dis. 2012, 2012, 649456. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.; Kalev, O.; Mehrabian, S.; Traykov, L.; Raycheva, M.; Kanakis, D.; Drineas, P.; Lutz, M.I.; Strobel, T.; Penz, T.; et al. Familial early-onset dementia with complex neuropathologic phenotype and genomic background. Neurobiol. Aging 2016, 42, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Alemi, M.; Silva, S.C.; Santana, I.; Cardoso, I. Transthyretin stability is critical in assisting β amyloid clearance—Relevance of transthyretin stabilization in Alzheimer’s disease. CNS Neurosci. Ther. 2017, 23, 605–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giao, T.; Saavedra, J.; Cotrina, E.; Quintana, J.; Llop, J.; Arsequell, G.; Cardoso, I. Undiscovered roles for Transthyretin: From a transporter protein to a new therapeutic target for Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 2075. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Masliah, E.; Reixach, N.; Buxbaum, J.N. Neuronal production of transthyretin in human and murine Alzheimer’s disease: Is it protective? J. Neurosci. 2011, 31, 12483–12490. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Li, Y.; Hu, Y.; Chen, C.; Zhou, Y.; Tao, Y.; Guo, M.; Qin, N.; Xu, L. MicroRNAs expression profile in CCR6(+) regulatory T cells. Peer J. 2014, 2, e575. [Google Scholar] [CrossRef] [Green Version]
- GeneCards. The Human Gene Database: Synaptotagmin 11. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=SYT11 (accessed on 31 May 2021).
- KEGG-Derived PI3K-Akt Signaling Pathway. Available online: https://www.genome.jp/kegg-bin/show_pathway?mmu04151 (accessed on 2 September 2020).
- Fragoso-Loyo, H.; Atisha-Fregoso, Y.; Nunez-Alvarez, C.A.; Llorente, L. Utility of TWEAK to assess neuropsychiatric disease activity in systemic lupus erhytematosus. Lupus 2016, 25, 364–369. [Google Scholar] [CrossRef]
- Mackay, M.; Tang, C.C.; Vo, A. Advanced neuroimaging in neuropsychiatric systemic lupus erythematosus. Curr. Opin. Neurol. 2020, 33, 353–361. [Google Scholar] [CrossRef]
- Chi, J.M.; Mackay, M.; Hoang, A.; Cheng, K.; Aranow, C.; Ivanidze, J.; Volpe, B.; Diamond, B.; Sanelli, P.C. Alterations in blood-brain barrier permeability in patients with Systemic Lupus Erythematosus. AJNR Am. J. Neuroradiol. 2019, 40, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Rodriguez, R.D.; Suemoto, C.K.; et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef]
- Tomita, M.; Holman, B.J.; Santoro, T.J. Aberrant cytokine gene expression in the hippocampus in murine systemic lupus erythematosus. Neurosci. Lett. 2001, 302, 129–132. [Google Scholar] [CrossRef]
- Tomita, M.; Holman, B.J.; Williams, L.S.; Pang, K.C.; Santoro, T.J. Cerebellar dysfunction is associated with overexpression of proinflammatory cytokine genes in lupus. J. Neurosci. Res. 2001, 64, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kowal, C.; DeGiorgio, L.A.; Nakaoka, T.; Hetherington, H.; Huerta, P.T.; Diamond, B.; Volpe, B.T. Cognition and immunity; antibody impairs memory. Immunity 2004, 21, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Stock, A.D.; Der, E.; Gelb, S.; Huang, M.; Weidenheim, K.; Ben-Zvi, A.; Putterman, C. Tertiary lymphoid structures in the choroid plexus in neuropsychiatric lupus. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Stock, A.D.; Wen, J.; Doerner, J.; Herlitz, L.C.; Gulinello, M.; Putterman, C. Neuropsychiatric systemic lupus erythematosus persists despite attenuation of systemic disease in MRL/lpr mice. J. Neuroinflamm. 2015, 12, 205. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Doerner, J.; Chalmers, S.; Stock, A.; Wang, H.; Gullinello, M.; Shlomchik, M.J.; Putterman, C. B cell and/or autoantibody deficiency do not prevent neuropsychiatric disease in murine systemic lupus erythematosus. J. Neuroinflamm. 2016, 13, 73. [Google Scholar] [CrossRef] [Green Version]
- Simen, A.A.; Bordner, K.A.; Martin, M.P.; Moy, L.A.; Barry, L.C. Cognitive dysfunction with aging and the role of inflammation. Ther. Adv. Chronic. Dis. 2011, 2, 175–195. [Google Scholar] [CrossRef] [Green Version]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Carroni, M.; Nussbaum-Krammer, C.; Mogk, A.; Nillegoda, N.B.; Szlachcic, A.; Guilbride, D.L.; Saibil, H.R.; Mayer, M.P.; Bukau, B. Human Hsp70 disaggregase reverses Parkinson’s-linked α-synuclein amyloid fibrils. Mol. Cell 2015, 59, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Kakimura, J.; Kitamura, Y.; Takata, K.; Umeki, M.; Suzuki, S.; Shibagaki, K.; Taniguchi, T.; Nomura, Y.; Gebicke-Haerter, P.J.; Smith, M.A.; et al. Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins. FASEB J. 2002, 16, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Popiel, H.A.; Takeuchi, T.; Fujita, H.; Yamamoto, K.; Ito, C.; Yamane, H.; Muramatsu, S.; Toda, T.; Wada, K.; Nagai, Y. Hsp40 gene therapy exerts therapeutic effects on polyglutamine disease mice via a non-cell autonomous mechanism. PLoS ONE 2012, 7, e51069. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Suzuki, M.; Fujikake, N.; Popiel, H.A.; Kikuchi, H.; Futaki, S.; Wada, K.; Nagai, Y. Intercellular chaperone transmission via exosomes contributes to maintenance of protein homeostasis at the organismal level. Proc. Natl. Acad. Sci. USA 2015, 112, E2497–E2506. [Google Scholar] [CrossRef] [Green Version]
- GeneCards. The Human Gene Database: SYNE2. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=SYNE2 (accessed on 31 May 2021).
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Abeta toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 2681–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K.; et al. Transthyretin sequesters amyloid β protein and prevents amyloid formation. Proc. Natl. Acad. Sci. USA 1994, 91, 8368–8372. [Google Scholar] [CrossRef] [Green Version]
- Trysberg, E.; Hoglund, K.; Svenungsson, E.; Blennow, K.; Tarkowski, A. Decreased levels of soluble amyloid β-protein precursor and β-amyloid protein in cerebrospinal fluid of patients with systemic lupus erythematosus. Arthritis Res. Ther. 2004, 6, R129–R136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Tschopp, O.; Yang, Z.Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential role of protein kinase B γ (PKB γ/Akt3) in postnatal brain development but not in glucose homeostasis. Development 2005, 132, 2943–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, R.M.; Cho, H.; Roovers, K.; Shineman, D.W.; Mizrahi, M.; Forman, M.S.; Lee, V.M.; Szabolcs, M.; de Jong, R.; Oltersdorf, T.; et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell. Biol. 2005, 25, 1869–1878. [Google Scholar] [CrossRef] [Green Version]
- Flores, A.I.; Narayanan, S.P.; Morse, E.N.; Shick, H.E.; Yin, X.; Kidd, G.; Avila, R.L.; Kirschner, D.A.; Macklin, W.B. Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. 2008, 28, 7174–7183. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B., 3rd; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB β). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yan, P.; Gong, H.; Zuo, L.; Shi, Y.; Guo, J.; Guo, R.; Xie, J.; Li, B. TWEAK protects cardiomyocyte against apoptosis in a PI3K/AKT pathway dependent manner. Am. J. Transl. Res. 2016, 8, 3848–3860. [Google Scholar]
- Dogra, C.; Changotra, H.; Wedhas, N.; Qin, X.; Wergedal, J.E.; Kumar, A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 2007, 21, 1857–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Xu, M.; Yin, X.; Guo, H.; Zhang, B.; Wang, Y.; Xiao, J.; Zou, X.; Zhang, M.; Zhuge, Y. TWEAK promotes hepatic stellate cell migration through activating EGFR/Src and PI3K/AKT pathways. Cell Biol. Int. 2019, 44, 278–285. [Google Scholar] [CrossRef]
- Xu, R.D.; Feng, F.; Yu, X.S.; Liu, Z.D.; Lao, L.F. miR-149-5p inhibits cell growth by regulating TWEAK/Fn14/PI3K/AKT pathway and predicts favorable survival in human osteosarcoma. Int J. Immunopathol. Pharmacol. 2018, 32, 2058738418786656. [Google Scholar] [CrossRef] [PubMed]
- Fortin, S.P.; Ennis, M.J.; Savitch, B.A.; Carpentieri, D.; McDonough, W.S.; Winkles, J.A.; Loftus, J.C.; Kingsley, C.; Hostetter, G.; Tran, N.L. Tumor necrosis factor-like weak inducer of apoptosis stimulation of glioma cell survival is dependent on Akt2 function. Mol. Cancer Res. 2009, 7, 1871–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Description | MRL/lpr | MRL/+ | Fn14ko |
|---|---|---|---|---|
| Dnajc28 | DnaJ (Hsp40) homolog, subfamily C, member 28 | 334.06 | 0.17 | 0.09 |
| Syne2 | synaptic nuclear envelope 2 | 316.02 | 0.18 | 0.09 |
| Rpp38 | ribonuclease P/MRP 38 subunit (human) | 314.63 | 1.03 | 1.31 |
| Cox7b | cytochrome c oxidase subunit VIIb | 311.86 | 23.40 | 31.39 |
| Kctd10 | potassium channel tetramerisation domain containing 10 | 307.63 | 0.32 | 0.09 |
| Tle6 | transducin-like enhancer of split 6, homolog of Drosophila E | 307.41 | 0.37 | 0.54 |
| Pcnx4 | pecanex homolog 4 | 306.69 | 1.26 | 1.81 |
| Dlx5 | distal-less homeobox 5 | 302.19 | 0.48 | 0.14 |
| Clec14a | C-type lectin domain family 14, member a | 300.99 | 0.21 | 0.14 |
| Coq6 | coenzyme Q6 homolog (yeast) | 290.92 | 2.95 | 3.18 |
| Cradd | CASP2 and RIPK1 domain containing adaptor with death domain | 289.92 | 0.40 | 0.60 |
| Ppp2ca | protein phosphatase 2 (formerly 2A), catalytic subunit, α isoform | 289.29 | 12.08 | 19.16 |
| Gps1 | G protein pathway suppressor 1 | 282.09 | 19.02 | 21.20 |
| Zfp455 | zinc finger protein 455 | 276.70 | 0.20 | 0.12 |
| Donson | downstream neighbor of SON | 267.23 | 0.83 | 1.36 |
| Tmie | transmembrane inner ear | 265.98 | 0.43 | 0.32 |
| Akt2 | thymoma viral proto-oncogene 2 | 261.94 | 4.09 | 7.64 |
| Deaf1 | deformed epidermal autoregulatory factor 1 (Drosophila) | 259.89 | 1.35 | 0.82 |
| Eif2s1 | eukaryotic translation initiation factor 2, subunit 1 α | 258.35 | 0.55 | 0.84 |
| Dnajc14 | DnaJ (Hsp40) homolog, subfamily C, member 14 | 256.52 | 0.77 | 0.63 |
| Gene | Description | MRL/lpr vs. MRL/+ | MRL/lpr vs. Fn14ko | ||
|---|---|---|---|---|---|
| x | WIR | x | WIR | ||
| Nrtn | neurturin | −17.12 | −1032.44 | −1.11 | −0.24 |
| Dnajc28 | DnaJ (Hsp40) homolog, subfamily C, member 28 | 1979.55 | 203.31 | 3817.12 | 203.39 |
| Syne2 | synaptic nuclear envelope 2 | 1714.27 | 192.30 | 3672.93 | 192.40 |
| Rpp38 | ribonuclease P/MRP 38 subunit (human) | 304.48 | 190.96 | 241.04 | 190.68 |
| Tle6 | transducin-like enhancer of split 6, homolog of Drosophila E(spl) | 830.65 | 187.22 | 572.06 | 187.05 |
| Kctd10 | potassium channel tetramerisation domain containing 10 | 950.07 | 187.04 | 3585.62 | 187.28 |
| Dlx5 | distal-less homeobox 5 | 635.75 | 183.64 | 2170.24 | 183.99 |
| Clec14a | C-type lectin domain family 14, member a | 1421.76 | 183.14 | 2104.76 | 183.21 |
| Cox7b | cytochrome c oxidase subunit VIIb | 13.33 | 178.48 | 9.93 | 170.27 |
| Cradd | CASP2 and RIPK1 domain containing adaptor with death domain | 719.92 | 176.35 | 484.24 | 176.15 |
| Coq6 | coenzyme Q6 homolog (yeast) | 98.49 | 175.50 | 91.40 | 175.27 |
| Ppp2ca | protein phosphatase 2 (formerly 2A), catalytic subunit, α isoform | 23.95 | 169.92 | 15.10 | 162.69 |
| Zfp455 | zinc finger protein 455 | 1373.60 | 168.35 | 2255.80 | 168.43 |
| Donson | downstream neighbor of SON | 322.96 | 162.56 | 196.84 | 162.02 |
| Tmie | transmembrane inner ear | 618.70 | 161.70 | 818.52 | 161.81 |
| Gps1 | G protein pathway suppressor 1 | 14.83 | 160.90 | 13.31 | 158.67 |
| Akt2 | thymoma viral proto-oncogene 2 | 64.02 | 158.94 | 34.28 | 155.29 |
| Deaf1 | deformed epidermal autoregulatory factor 1 (Drosophila) | 193.09 | 157.27 | 316.93 | 157.80 |
| Eif2s1 | eukaryotic translation initiation factor 2, subunit 1 α | 472.78 | 157.09 | 307.81 | 156.79 |
| Dnajc14 | DnaJ (Hsp40) homolog, subfamily C, member 14 | 332.10 | 155.60 | 410.38 | 155.75 |
| Arrdc1 | arrestin domain containing 1 | 461.59 | 155.23 | 406.91 | 155.15 |
| Nkap | NFKB activating protein | −23.74 | −146.42 | 1.12 | 0.04 |
| Sall1 | sal-like 1 (Drosophila) | 515.64 | 142.36 | 977.24 | 142.58 |
| Rnmtl1 | RNA methyltransferase like 1 | 286.57 | 141.15 | 314.45 | 141.22 |
| Ybx2 | Y box protein 2 | 799.94 | 139.73 | 707.63 | 139.70 |
| Arl4a | ADP-ribosylation factor-like 4A | 203.58 | 139.32 | 117.37 | 138.48 |
| Tbc1d19 | TBC1 domain family, member 19 (Tbc1d19), mRNA [NM_144517] | 259.29 | 136.98 | 213.37 | 136.79 |
| Lrrc2 | leucine rich repeat containing 2 | 1145.66 | 135.04 | 1901.54 | 135.11 |
| Gpr183 | G protein-coupled receptor 183 | 1187.49 | 131.43 | 2459.92 | 131.53 |
| Abcc3 | ATP-binding cassette, sub-family C (CFTR/MRP), member 3 | 988.46 | 131.05 | 1673.97 | 131.14 |
| Ccdc124 | coiled-coil domain containing 124 | 16.46 | 121.35 | 13.70 | 118.72 |
| Rgl1 | ral guanine nucleotide dissociation stimulator-like 1 | 113.72 | 118.88 | 182.93 | 119.55 |
| Mki67 | antigen identified by monoclonal antibody Ki 67 | 903.22 | 101.46 | 977.16 | 101.48 |
| Hba-a1 | hemoglobin α, adult chain 1 | 3.69 | 91.05 | 1.82 | 54.31 |
| Hba-a2 | hemoglobin α, adult chain 2 | 2.54 | 90.32 | 1.77 | 64.14 |
| Tank | TRAF family member-associated Nf-kappa B activator | 381.55 | 87.10 | 407.55 | 87.12 |
| Slc10a1 | solute carrier family 10 (sodium/bile acid cotransporter family), member 1 | 1.82 | 83.41 | 1.82 | 83.18 |
| H2-Gs10 | MHC class I like protein GS10 | 10.38 | 82.27 | 9.02 | 80.23 |
| Rnf168 | ring finger protein 168 | 87.22 | 80.35 | 106.75 | 80.63 |
| Elk1 | ELK1, member of ETS oncogene family | 1.59 | 65.37 | 1.75 | 75.96 |
| Exosc2 | exosome component 2 | 1.74 | 62.55 | 1.39 | 39.39 |
| Cxcl11 | chemokine (C-X-C motif) ligand 11 | 2.82 | 61.18 | 2.68 | 59.47 |
| Ttr | transthyretin | −2.00 | −52.38 | −1.76 | −40.27 |
| Kif5a | kinesin family member 5A | 1.69 | 52.29 | 1.30 | 26.92 |
| Hccs | holocytochrome c synthetase | 3.37 | 50.04 | 3.14 | 48.37 |
| Zfp628 | zinc finger protein 628 | −1.34 | −47.43 | 1.36 | 28.96 |
| Atp6v0c | ATPase, H+ transporting, lysosomal V0 subunit C | −1.40 | −43.10 | −1.18 | −11.73 |
| Gla | galactosidase, α | 2.04 | 41.87 | 1.61 | 31.24 |
| Psap | prosaposin | −1.66 | −41.72 | −1.16 | −7.03 |
| Dcc | deleted in colorectal carcinoma | 2.30 | 41.29 | 3.40 | 52.26 |
| Oxt | oxytocin | −16.11 | −39.14 | 1.02 | 0.00 |
| Tubb2a | tubulin, β 2A | −1.35 | −38.27 | −1.41 | −36.56 |
| Ddn | dendrin | −2.34 | −38.13 | −1.28 | −3.17 |
| Ptms | parathymosin | −1.44 | −37.91 | 1.19 | 8.51 |
| Acot7 | acyl-CoA thioesterase 7 | −1.71 | −35.06 | −1.15 | −3.35 |
| Ptgds | prostaglandin D2 synthase (brain) | 1.45 | 34.82 | 1.23 | 11.03 |
| Thy1 | thymus cell antigen 1, theta | −1.32 | −34.56 | 1.18 | 11.17 |
| Sparcl1 | SPARC-like 1 | 1.42 | 33.77 | 1.01 | 0.13 |
| Pkm2 | pyruvate kinase, muscle | −1.57 | −33.58 | −1.13 | −3.69 |
| Cox8a | cytochrome c oxidase, subunit VIIIa | 1.31 | 32.71 | 1.19 | 17.94 |
| Gene | Description | MRL/+ | MRL/lpr | Fn14ko |
|---|---|---|---|---|
| Tfb2m | transcription factor B2, mitochondrial | 234 | 3 | 9 |
| Hiatl1 | hippocampus abundant transcript-like 1 | 85 | 4 | 2 |
| Mettl11a | methyltransferase like 11A | 73 | 1 | 4 |
| Gipc1 | GIPC PDZ domain containing family, member 1 | 68 | 2 | 1 |
| Homez | homeodomain leucine zipper-encoding gene | 60 | 2 | 2 |
| Syt11 | synaptotagmin XI | 58 | 2 | 3 |
| Eef1g | eukaryotic translation elongation factor 1 γ | 54 | 4 | 2 |
| Brsk1 | BR serine/threonine kinase 1 | 53 | 2 | 2 |
| Gga2 | golgi associated, γ adaptin ear containing, ARF binding protein 2 | 50 | 1 | 2 |
| St3gal5 | ST3 β-galactoside α-2,3-sialyltransferase 5 | 49 | 1 | 2 |
| Dusp22 | dual specificity phosphatase 22 | 46 | 2 | 1 |
| Avl9 | AVL9 homolog | 42 | 2 | 3 |
| Foxq1 | forkhead box Q1 | 42 | 1 | 1 |
| Lmna | lamin A | 42 | 3 | 1 |
| Anpep | alanyl (membrane) aminopeptidase | 41 | 1 | 1 |
| Xrcc4 | X-ray repair complementing defective repair in Chinese hamster cells 4 | 4 | 43 | 1 |
| Tubb4 | tubulin, β 4 | 2 | 43 | 2 |
| Arhgap23 | Rho GTPase activating protein 23 | 2 | 36 | 3 |
| Ecd | ecdysoneless homolog | 5 | 35 | 5 |
| Cacfd1 | calcium channel flower domain containing 1 | 5 | 29 | 2 |
| Nacad | NAC α domain containing | 10 | 26 | 2 |
| Pnldc1 | poly(A)-specific ribonuclease (PARN)-like domain containing 1 | 3 | 23 | 2 |
| Mrpl40 | mitochondrial ribosomal protein L40 | 1 | 22 | 3 |
| Serf2 | small EDRK-rich factor 2 | 6 | 22 | 2 |
| Rpa2 | replication protein A2 | 7 | 22 | 3 |
| Unc79 | unc-79 homolog | 4 | 20 | 5 |
| Pogk | pogo transposable element with KRAB domain | 8 | 19 | 3 |
| Gabpb1 | GA repeat binding protein, β 1 | 2 | 18 | 5 |
| Mad2l2 | MAD2 mitotic arrest deficient-like 2 | 3 | 18 | 10 |
| Ccdc101 | coiled-coil domain containing 101 | 2 | 18 | 2 |
| Card19 | caspase recruitment domain family, member 19 | 2 | 2 | 27 |
| Ppfia1 | protein tyrosine phosphatase, receptor type, f polypeptide, interacting protein (liprin), α 1 | 6 | 5 | 26 |
| Map4k5 | mitogen-activated protein kinase kinase kinase kinase 5 | 2 | 2 | 25 |
| Ddx59 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 59 | 2 | 1 | 25 |
| Fbxo25 | F-box protein 25 | 2 | 1 | 21 |
| Rg9mtd3 | RNA (guanine-9-) methyltransferase domain containing 3 | 3 | 2 | 20 |
| Iscu | IscU iron-sulfur cluster scaffold homolog, nuclear gene encoding mitochondrial protein | 3 | 3 | 20 |
| Gm14407 | 60S ribosomal protein L27a pseudogene | 3 | 2 | 19 |
| Riok2 | RIO kinase 2 | 3 | 5 | 19 |
| Slc25a47 | solute carrier family 25, member 47 | 3 | 2 | 19 |
| Mapre3 | microtubule-associated protein, RP/EB family, member 3 | 9 | 2 | 19 |
| Pomc | pro-opiomelanocortin-α | 3 | 5 | 18 |
| Polr1e | polymerase (RNA) I polypeptide E | 6 | 3 | 18 |
| Olfr1347 | olfactory receptor 1347 | 3 | 4 | 17 |
| Tor2a | torsin family 2, member A | 9 | 1 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobas, D.A.; Wen, J.; Iacobas, S.; Putterman, C.; Schwartz, N. TWEAKing the Hippocampus: The Effects of TWEAK on the Genomic Fabric of the Hippocampus in a Neuropsychiatric Lupus Mouse Model. Genes 2021, 12, 1172. https://doi.org/10.3390/genes12081172
Iacobas DA, Wen J, Iacobas S, Putterman C, Schwartz N. TWEAKing the Hippocampus: The Effects of TWEAK on the Genomic Fabric of the Hippocampus in a Neuropsychiatric Lupus Mouse Model. Genes. 2021; 12(8):1172. https://doi.org/10.3390/genes12081172
Chicago/Turabian StyleIacobas, Dumitru A., Jing Wen, Sanda Iacobas, Chaim Putterman, and Noa Schwartz. 2021. "TWEAKing the Hippocampus: The Effects of TWEAK on the Genomic Fabric of the Hippocampus in a Neuropsychiatric Lupus Mouse Model" Genes 12, no. 8: 1172. https://doi.org/10.3390/genes12081172
APA StyleIacobas, D. A., Wen, J., Iacobas, S., Putterman, C., & Schwartz, N. (2021). TWEAKing the Hippocampus: The Effects of TWEAK on the Genomic Fabric of the Hippocampus in a Neuropsychiatric Lupus Mouse Model. Genes, 12(8), 1172. https://doi.org/10.3390/genes12081172