
Extensive Changes in Transcription Dynamics Reflected on Alternative Splicing Events in Systemic Lupus Erythematosus Patients



Abstract
:1. Introduction
2. Materials and Methods
2.1. SLE RNASeq Data Integration and Patient Stratification
2.2. Differential Splicing Analysis
2.3. Prediction of Nonsense Mediated Decay
2.4. Differential Gene Expression Analysis
3. Results
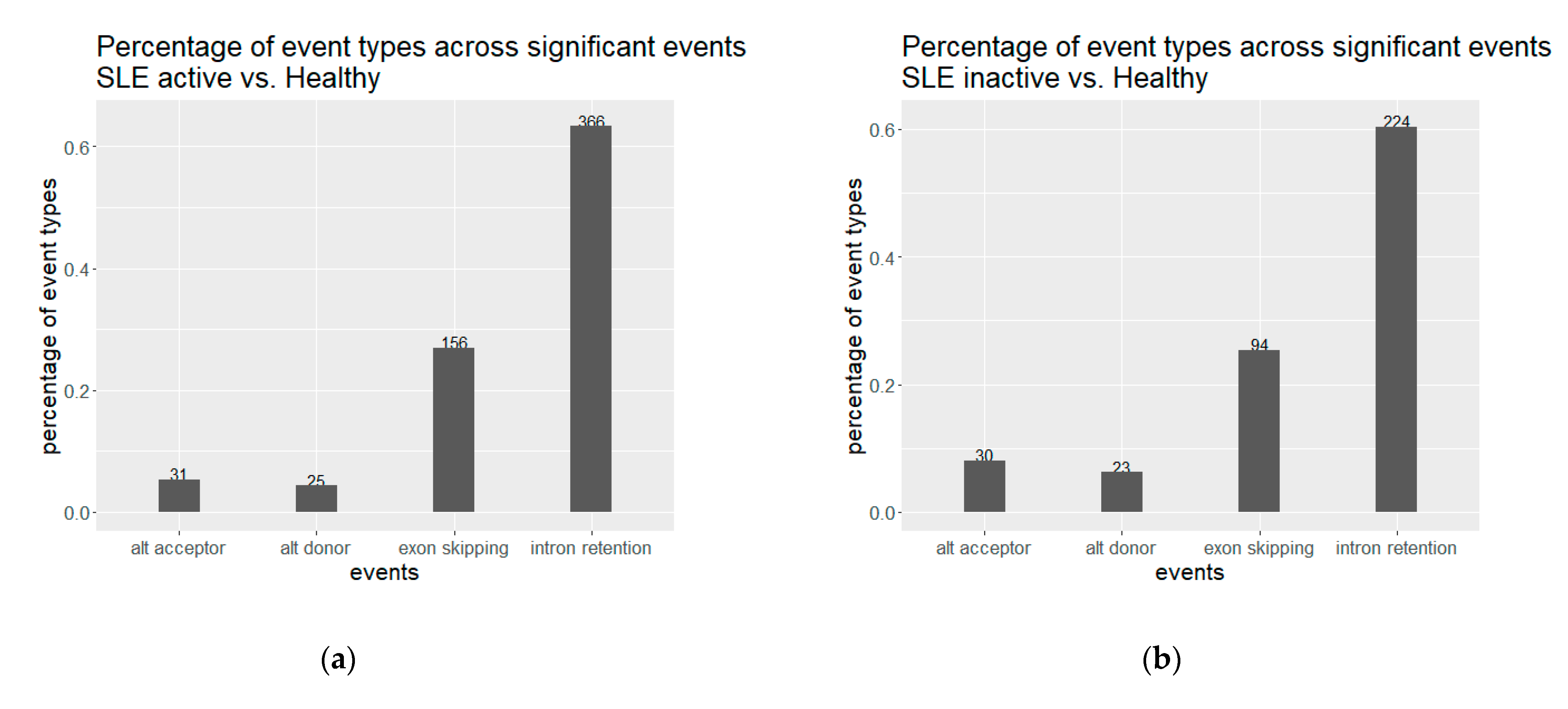
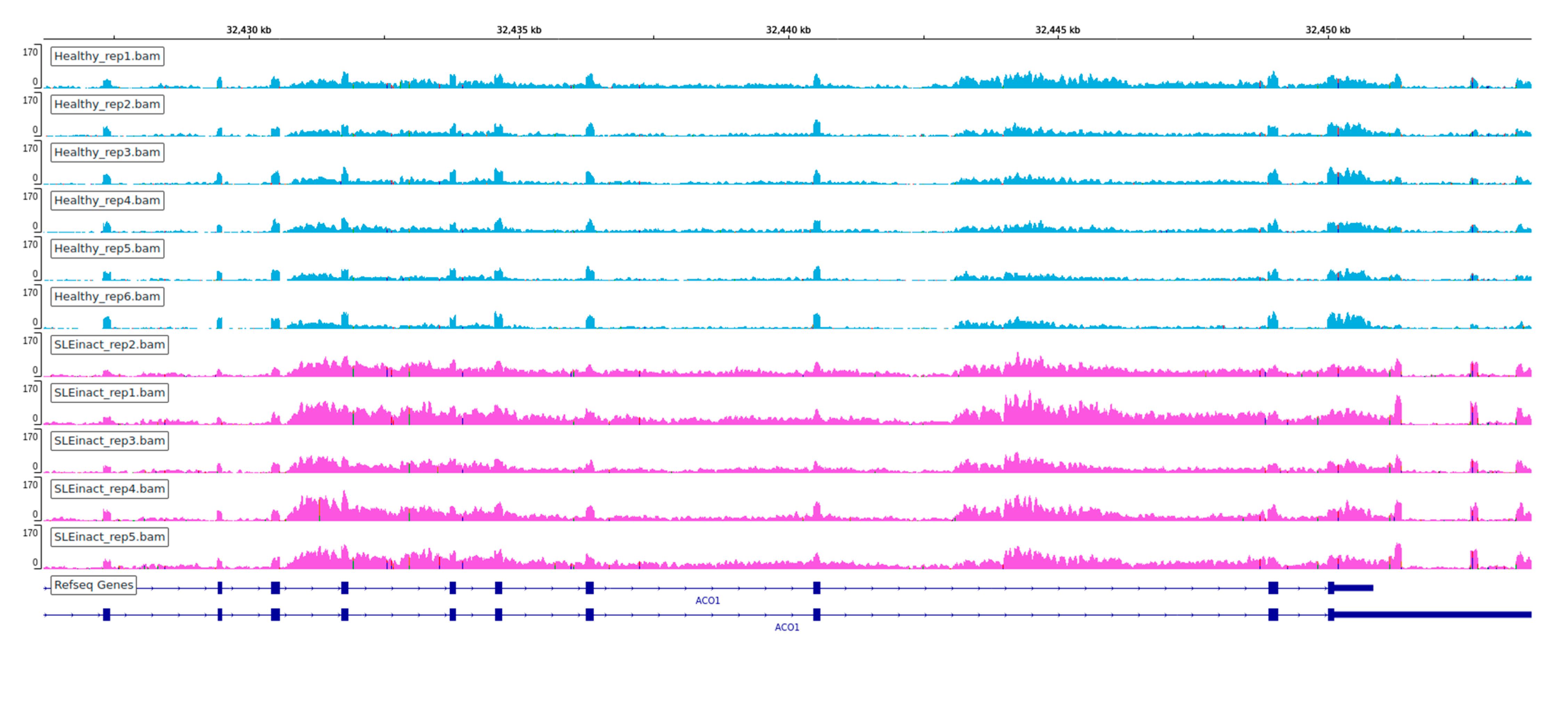
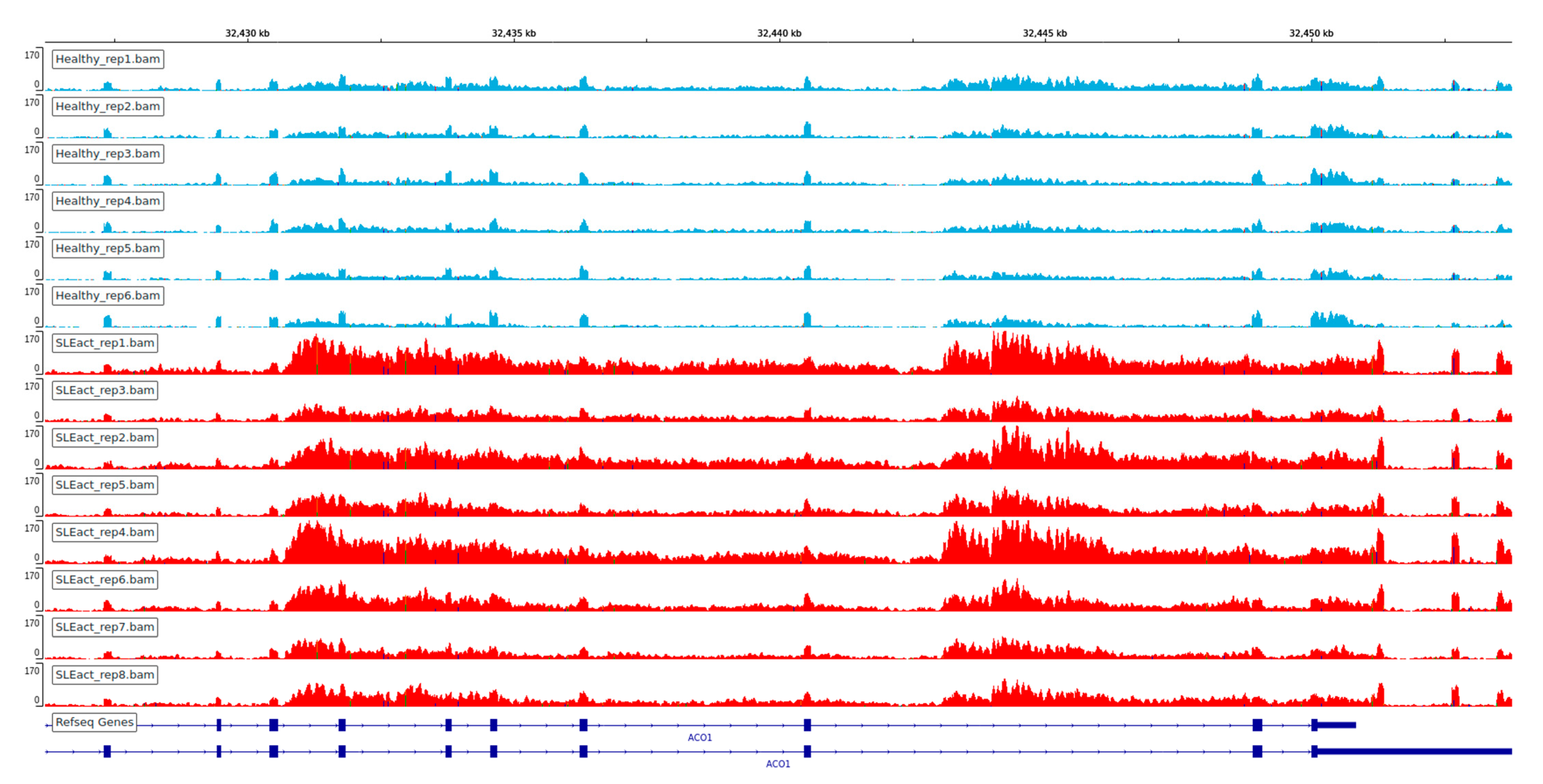
3.1. Extensive Perturbation of Splicing and Predominance of Intron Retention Events
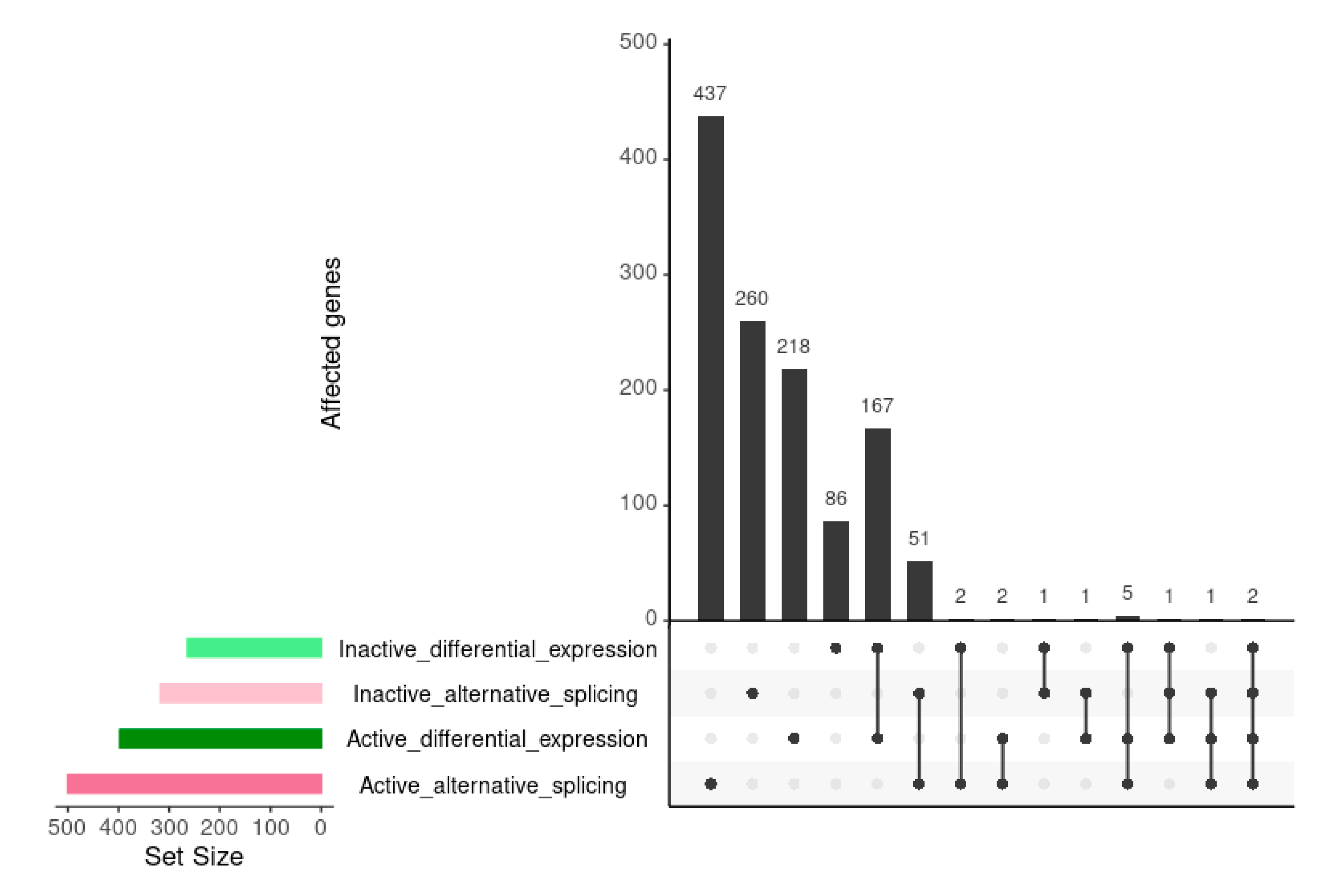
3.2. Relationship between Differentially Expressed Genes and Alternative Splicing Events
3.2.1. Alternative Splicing and Differential Expression Involve Different Genes
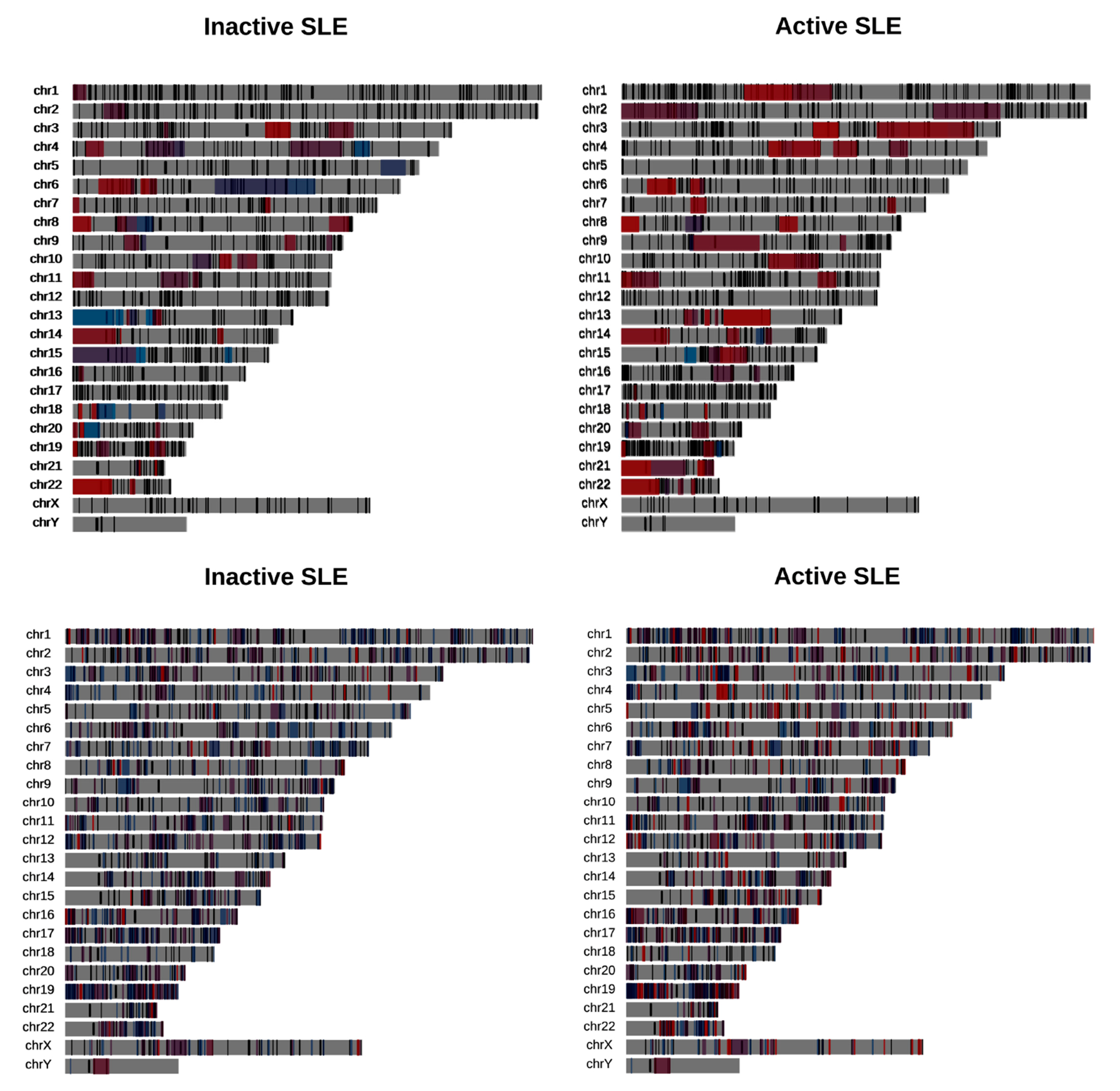
3.2.2. Alternative Splicing Takes Place in Gene Co-Expression Rather than Gene Deregulation Domains
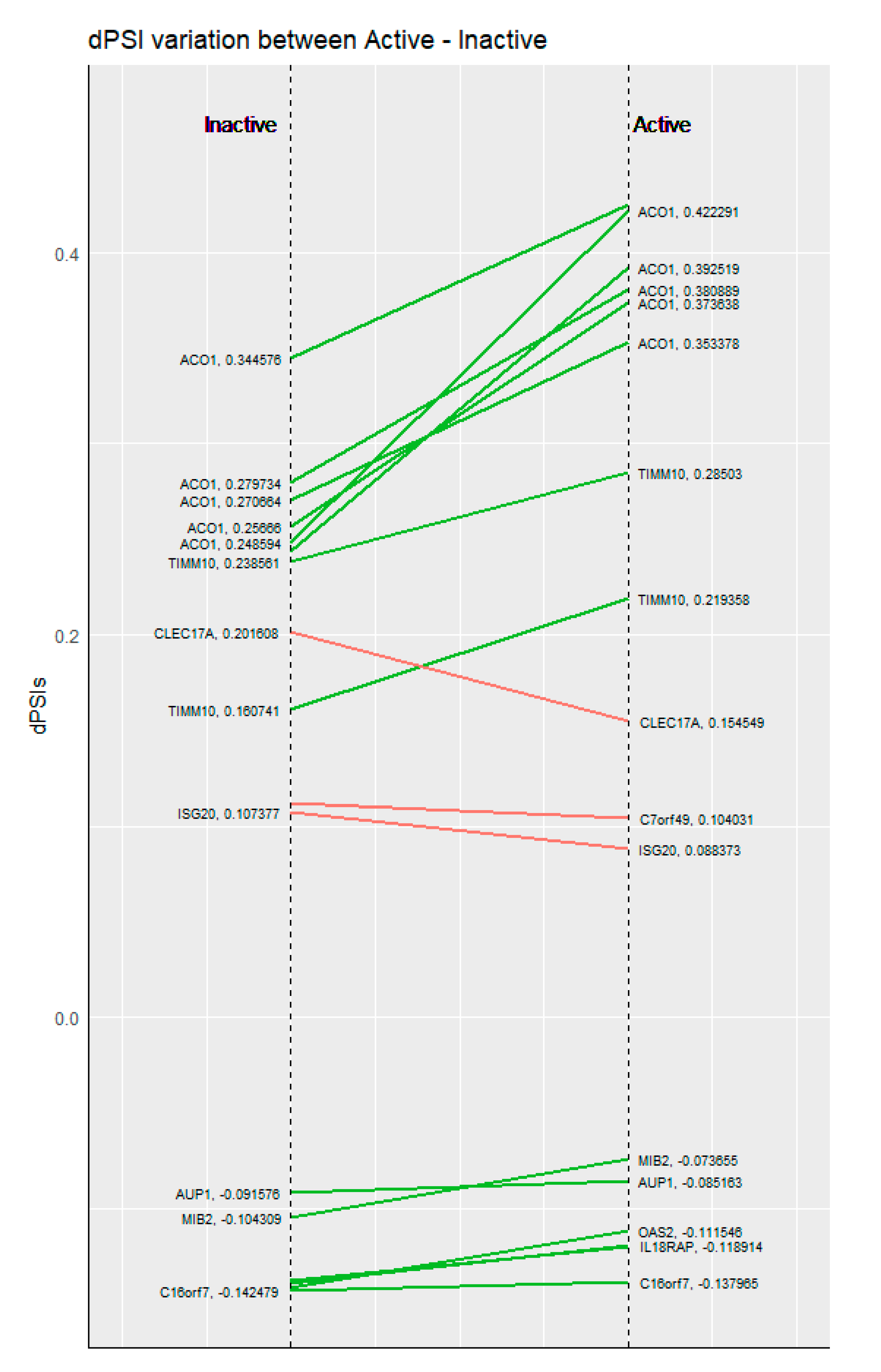
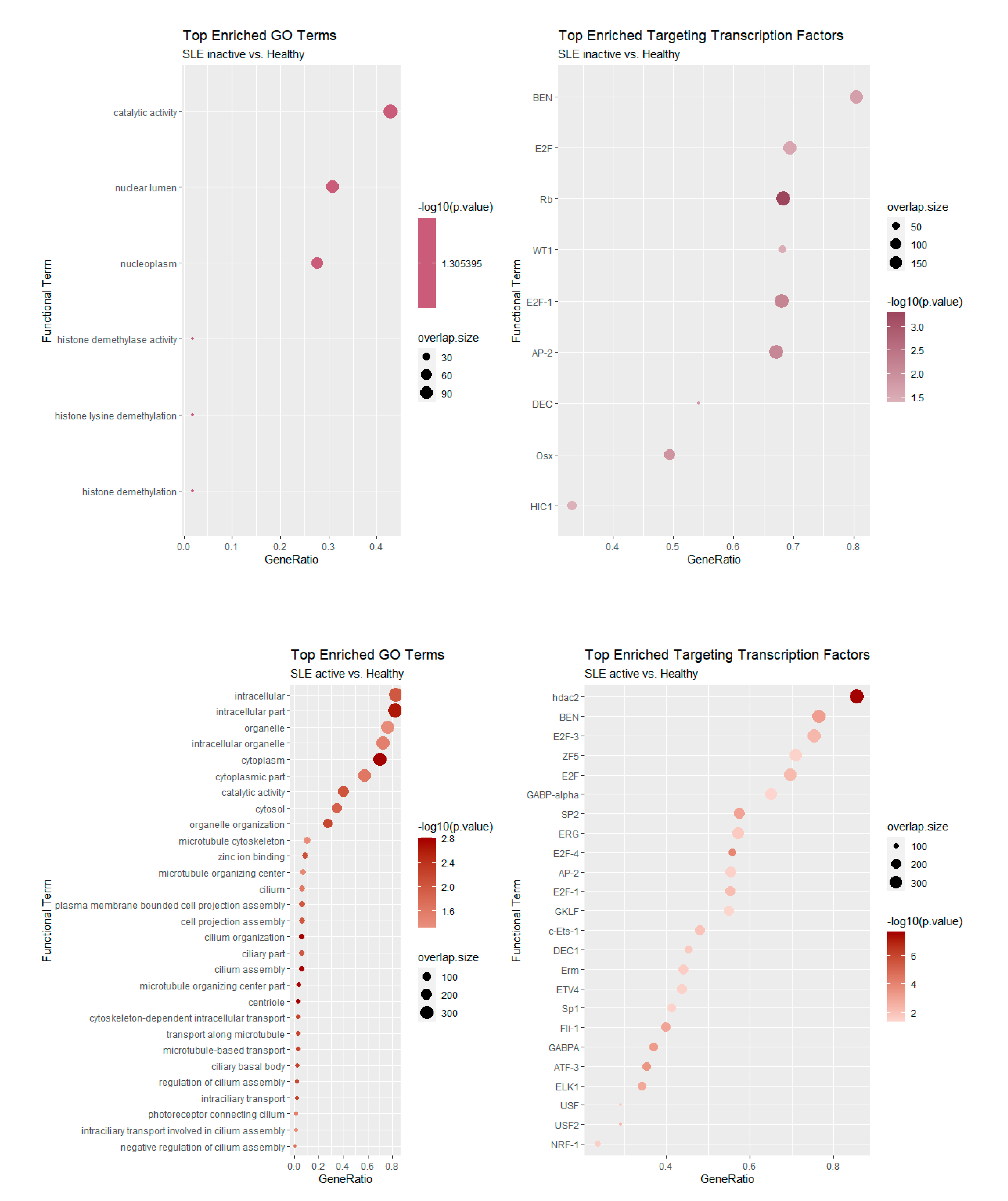
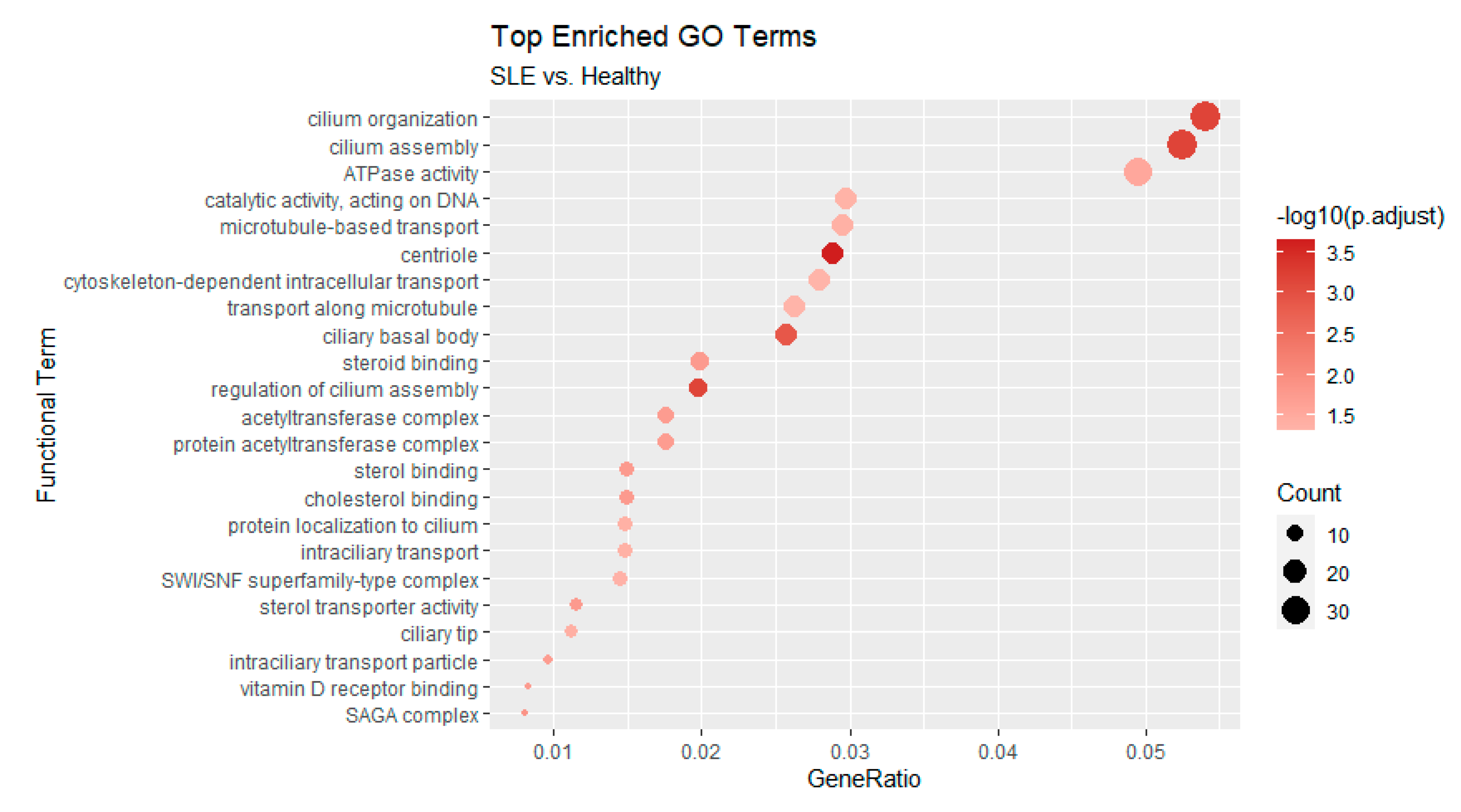
3.3. Functional Enrichment of Alternative Splicing Suggests Previously Unreported Genes and Functions in the Context of SLE
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Magen, A.; Ast, G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007, 35, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galante, P.A.F.; Sakabe, N.J.; Kirschbaum-Slager, N.; de Souza, S.J. Detection and evaluation of intron retention events in the human transcriptome. RNA 2004, 10, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiller, M.; Platzer, M. Widespread and subtle: Alternative splicing at short-distance tandem sites. Trends Genet. 2008, 24, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Ramanouskaya, T.V.; Grinev, V.V. The determinants of alternative RNA splicing in human cells. Mol. Genet. Genom. 2017, 292, 1175–1195. [Google Scholar] [CrossRef] [PubMed]
- Maquat, L.E. When cells stop making sense: Effects of nonsense codons on RNA metabolism in vertebrate cells. RNA 1995, 1, 453–465. [Google Scholar] [PubMed]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Maquat, L.E. Nonsense-mediated mRNA decay: Splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004, 5, 89–99. [Google Scholar] [CrossRef]
- Lloyd, J.P. The evolution and diversity of the nonsense-mediated mRNA decay pathway. F1000Research 2018, 7, 1299. [Google Scholar] [CrossRef]
- Braunschweig, U.; Barbosa-Morais, N.L.; Pan, Q.; Nachman, E.N.; Alipanahi, B.; Gonatopoulos-Pournatzis, T.; Frey, B.; Irimia, M.; Blencowe, B.J. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014, 24, 1774–1786. [Google Scholar] [CrossRef]
- Ge, Y.; Porse, B.T. The functional consequences of intron retention: Alternative splicing coupled to NMD as a regulator of gene expression. BioEssays 2013, 36, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Monteuuis, G.; Wong, J.J.; Bailey, C.G.; Schmitz, U.; Rasko, J.E. The changing paradigm of intron retention: Regulation, ramifications and recipes. Nucleic Acids Res. 2019, 47, 11497–11513. [Google Scholar] [CrossRef]
- Dam, E.M.; Habib, T.; Chen, J.; Funk, A.; Glukhova, V.; Davis-Pickett, M.; Wei, S.; James, R.; Buckner, J.H.; Cerosaletti, K. The BANK1 SLE-risk variants are associated with alterations in peripheral B cell signaling and development in humans. Clin. Immunol. 2016, 173, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Mamegano, K.; Kuroki, K.; Miyashita, R.; Kusaoi, M.; Kobayashi, S.; Matsuta, K.; Maenaka, K.; Colonna, M.; Ozaki, S.; Hashimoto, H.; et al. Association of LILRA2 (ILT1, LIR7) splice site polymorphism with systemic lupus erythematosus and microscopic polyangiitis. Genes Immun. 2008, 9, 214–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odhams, C.A.; Cortini, A.; Chen, L.; Roberts, A.L.; Vinuela, A.; Buil, A.; Small, K.S.; Dermitzakis, E.T.; Morris, D.L.; Vyse, T.J.; et al. Mapping eQTLs with RNA-seq reveals novel susceptibility genes, noncoding RNAs and alternative-splicing events in systemic lupus erythematosus. Hum. Mol. Genet. 2017, 26, 1003–1017. [Google Scholar] [CrossRef] [Green Version]
- Panousis, N.I.; Bertsias, G.K.; Ongen, H.; Gergianaki, I.; Tektonidou, M.G.; Trachana, M.; Romano-Palumbo, L.; Bielser, D.; Howald, C.; Pamfil, C.; et al. Combined genetic and transcriptome analysis of patients with SLE: Distinct, targetable signatures for susceptibility and severity. Ann. Rheum. Dis. 2019, 78, 1079–1089. [Google Scholar] [CrossRef]
- Irimia, M.; Weatheritt, R.J.; Ellis, J.D.; Parikshak, N.N.; Gonatopoulos-Pournatzis, T.; Babor, M.; Quesnel-Vallières, M.; Tapial, J.; Raj, B.; O’Hanlon, D.; et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 2014, 59, 1511–1523. [Google Scholar] [CrossRef] [Green Version]
- Tapial, J.; Ha, K.C.; Sterne-Weiler, T.; Gohr, A.; Braunschweig, U.; Hermoso-Pulido, A.; Quesnel-Vallières, M.; Permanyer, J.; Sodaei, R.; Marquez, Y.; et al. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017, 27, 1759–1768. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Braunschweig, U.; Gonatopoulos-Pournatzis, T.; Weatheritt, R.J.; Hirsch, C.L.; Ha, K.C.; Radovani, E.; Nabeel-Shah, S.; Sterne-Weiler, T.; Wang, J.; et al. Multilayered control of alternative splicing regulatory networks by transcription factors. Mol. Cell 2017, 65, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A web server for functional enrichment analysis and conversions of gene lists. Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Papoudas, S.M.; Papanikolaou, N.; Nikolaou, C. Monitoring the prolonged Tnf stimulation in space and time with topological-functional networks. Comput. Struct. Biotechnol. J. 2020, 18, 220–229. [Google Scholar] [CrossRef]
- Ntasis, V.F.; Panousis, N.I.; Tektonidou, M.G.; Dermitzakis, E.T.; Boumpas, D.T.; Bertsias, G.K.; Nikolaou, C. Extensive fragmentation and re-organization of transcription in Systemic Lupus Erythematosus. Sci. Rep. 2020, 10, 16648. [Google Scholar] [CrossRef]
- Andreadis, C.; Nikolaou, C.; Fragiadakis, G.S.; Tsiliki, G.; Alexandraki, D. Rad9 interacts with Aft1 to facilitate genome surveillance in fragile genomic sites under non-DNA damage-inducing conditions in S. cerevisiae. Nucleic Acids Res. 2014, 42, 12650–12667. [Google Scholar] [CrossRef] [Green Version]
- Smart, A.C.; Margolis, C.A.; Pimentel, H.; He, M.X.; Miao, D.; Adeegbe, D.; Fugmann, T.; Wong, K.K.; Van Allen, E.M. Intron retention is a source of neoepitopes in cancer. Nat. Biotechnol. 2018, 36, 1056–1058. [Google Scholar] [CrossRef]
- Zheng, J.T.; Lin, C.X.; Fang, Z.Y.; Li, H.D. Intron Retention as a Mode for RNA-Seq Data Analysis. Front. Genet. 2020, 11, 586. [Google Scholar] [CrossRef] [PubMed]
- Rahhal, R.; Seto, E. Emerging roles of histone modifications and HDACs in RNA splicing. Nucleic Acids Res. 2019, 47, 4911–4926. [Google Scholar] [CrossRef] [Green Version]
- Garcia, B.A.; Busby, S.A.; Shabanowitz, J.; Hunt, D.F.; Mishra, N. Resetting the epigenetic histone code in the MRL-lpr/lpr mouse model of lupus by histone deacetylase inhibition. J. Proteome Res. 2005, 4, 2032–2042. [Google Scholar] [CrossRef]
- Szyf, M. Epigenetic therapeutics in autoimmune disease. Clin. Rev. Allergy Immunol. 2010, 39, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Wheatley, D.N. Primary Cilia in Normal and Pathological Tissues. Pathobiology 1995, 63, 222–238. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Otto, E. Cilia and centrosomes: A unifying pathogenic concept for cystic kidney disease? Nat. Rev. Genet. 2005, 6, 928–940. [Google Scholar] [CrossRef]
- Stephen, L.A.; ElMaghloob, Y.; McIlwraith, M.J.; Yelland, T.; Sanchez, P.C.; Roda-Navarro, P.; Ismail, S. The Ciliary Machinery Is Repurposed for T Cell Immune Synapse Trafficking of LCK. Dev. Cell 2018, 47, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Asano, Y.; Griffiths, G. Origins of the cytolytic synapse. Nat. Rev. Immunol. 2016, 16, 421–432. [Google Scholar] [CrossRef]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Irg1 catalyzes itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Number of Samples | Number of Subgroups |
|---|---|---|
| SLE active | 79 | 8 |
| SLE inactive | 46 | 5 |
| Healthy | 58 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papanikolaou, S.; Bertsias, G.K.; Nikolaou, C. Extensive Changes in Transcription Dynamics Reflected on Alternative Splicing Events in Systemic Lupus Erythematosus Patients. Genes 2021, 12, 1260. https://doi.org/10.3390/genes12081260
Papanikolaou S, Bertsias GK, Nikolaou C. Extensive Changes in Transcription Dynamics Reflected on Alternative Splicing Events in Systemic Lupus Erythematosus Patients. Genes. 2021; 12(8):1260. https://doi.org/10.3390/genes12081260
Chicago/Turabian StylePapanikolaou, Sofia, George K. Bertsias, and Christoforos Nikolaou. 2021. "Extensive Changes in Transcription Dynamics Reflected on Alternative Splicing Events in Systemic Lupus Erythematosus Patients" Genes 12, no. 8: 1260. https://doi.org/10.3390/genes12081260
APA StylePapanikolaou, S., Bertsias, G. K., & Nikolaou, C. (2021). Extensive Changes in Transcription Dynamics Reflected on Alternative Splicing Events in Systemic Lupus Erythematosus Patients. Genes, 12(8), 1260. https://doi.org/10.3390/genes12081260