
Chromosome-Level Genome Assemblies Expand Capabilities of Genomics for Conservation Biology

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic Data
2.2. Quality Control and Filtration of Data
2.3. Alignment and Variant Calling
2.4. Identification of X Chromosome, Autosomes, and Pseudoautosomal Region
2.5. Comparison of Heterozygosity in Autosomes, X Chromosome, and PAR
2.6. Heterozygosity Visualization
2.7. Mapping of Known STR Loci on Chromosome-Level Assemblies of Mustelid Species
3. Results
3.1. Evaluation of the Genome Assemblies
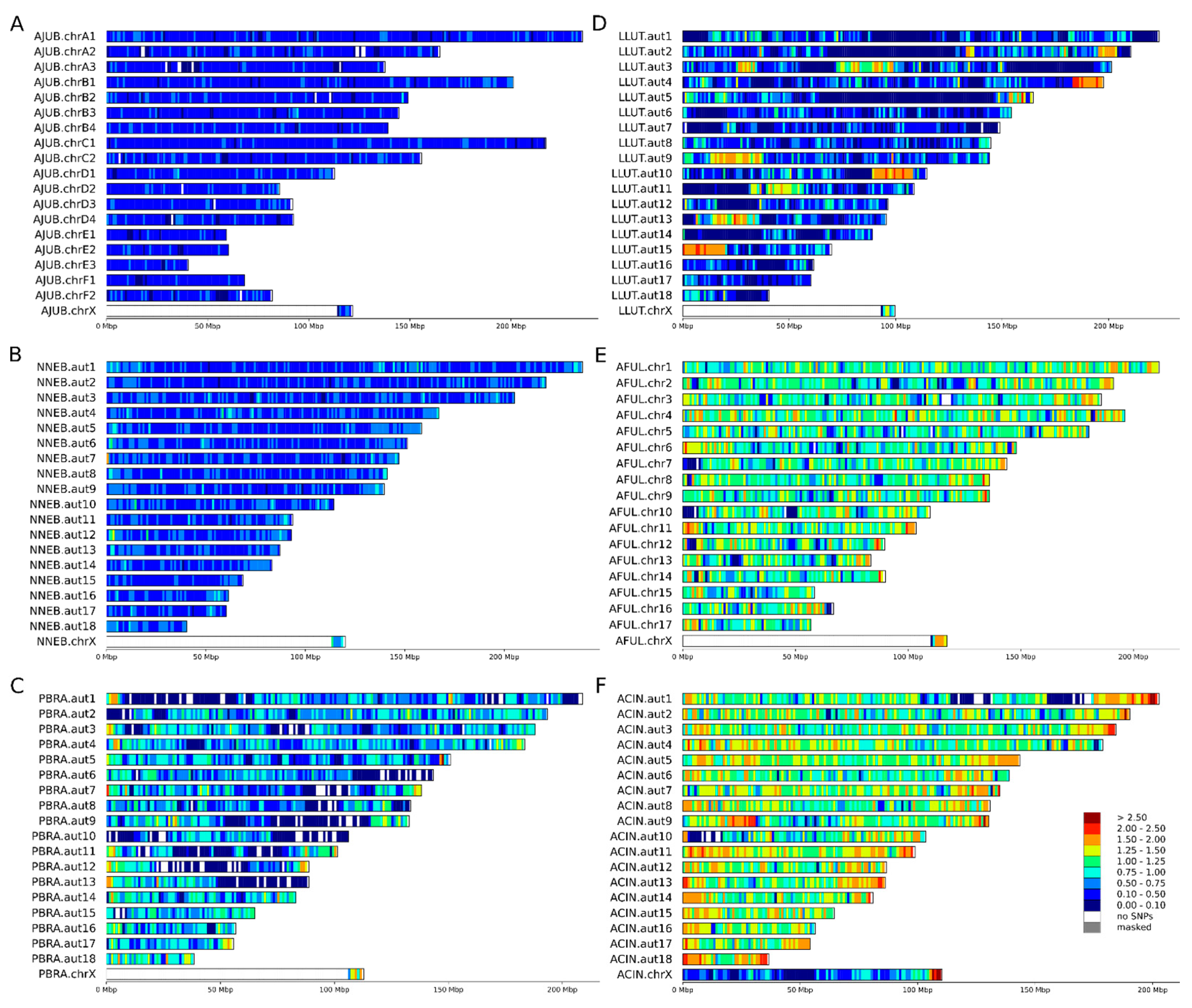
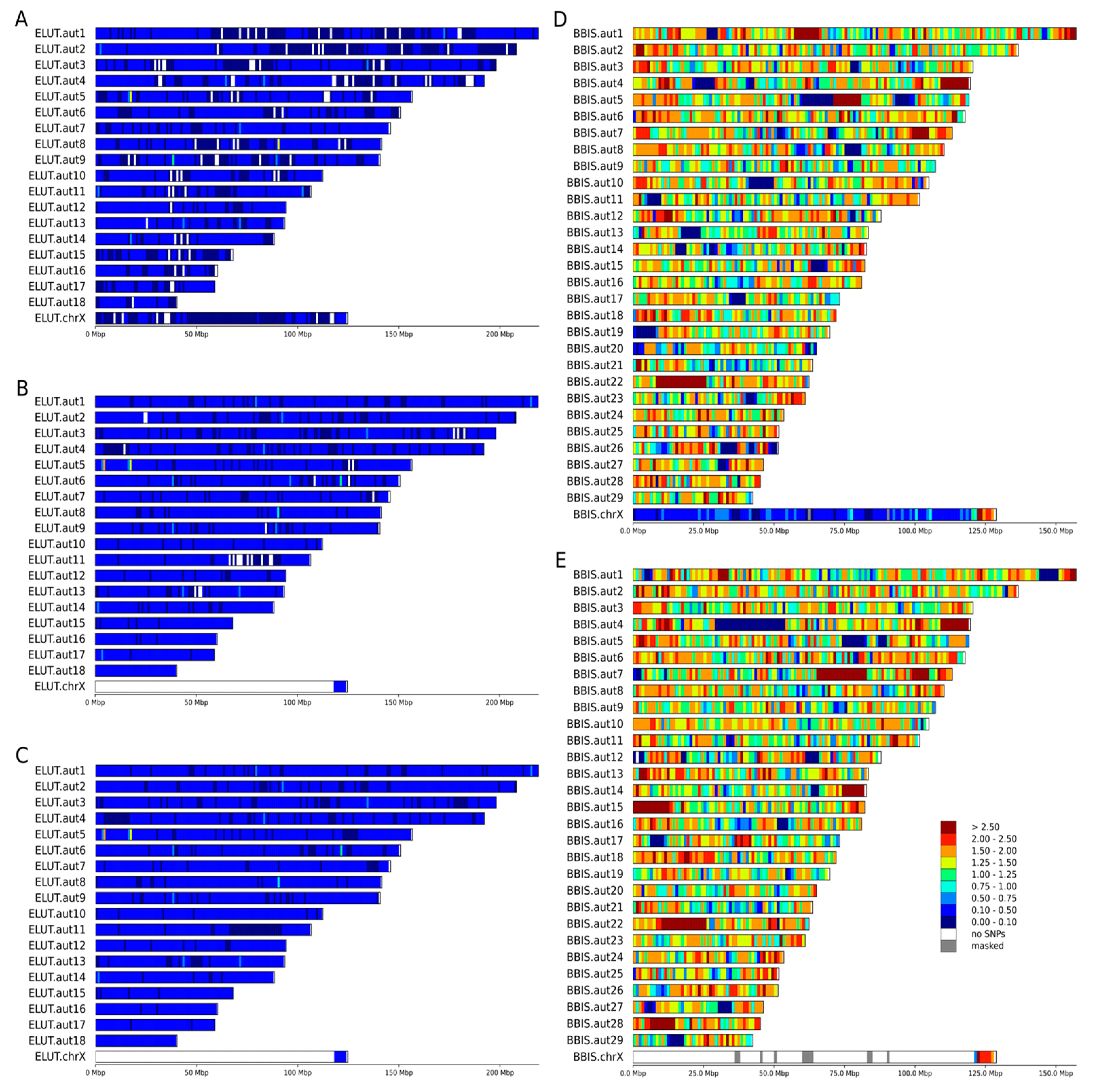
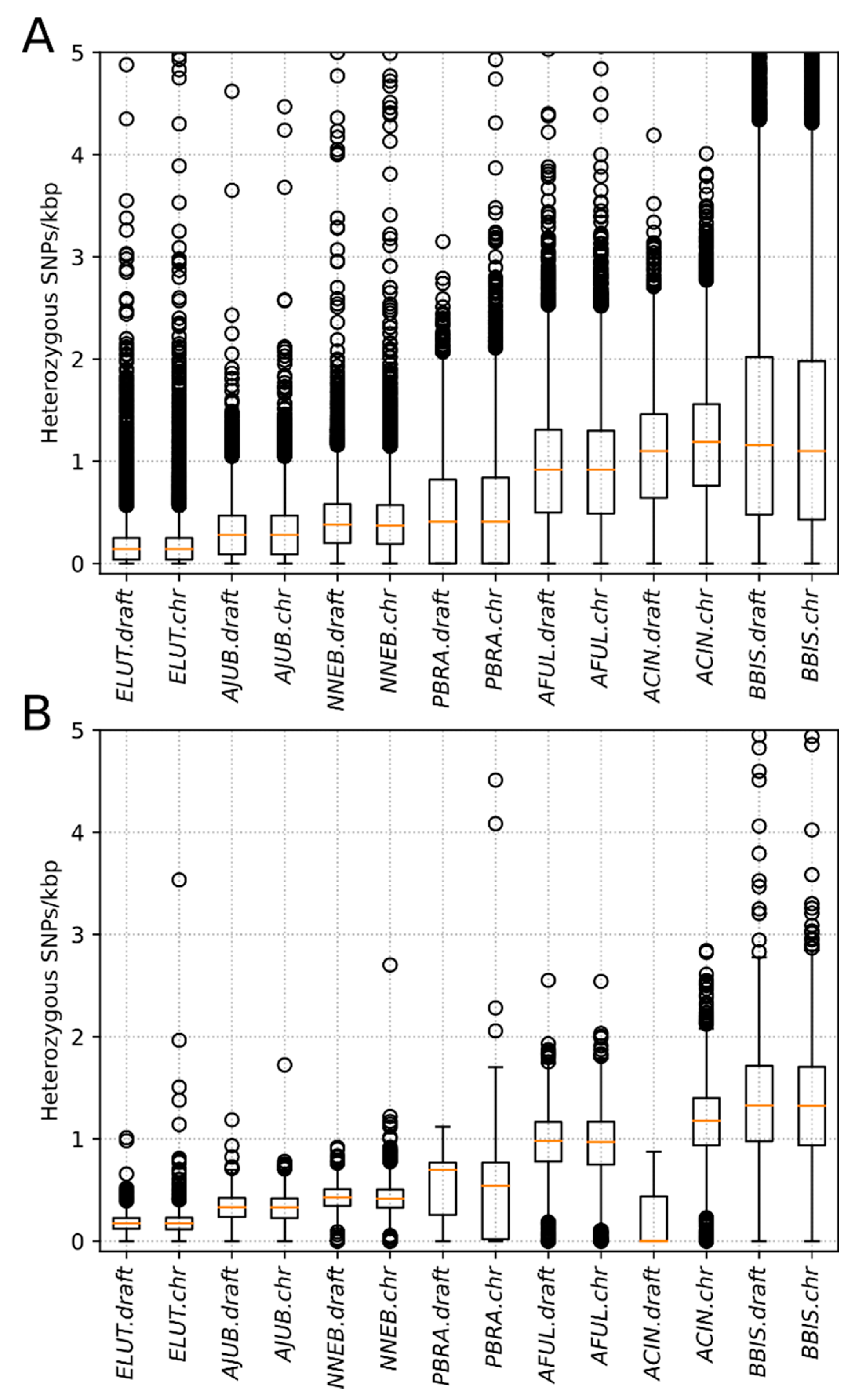
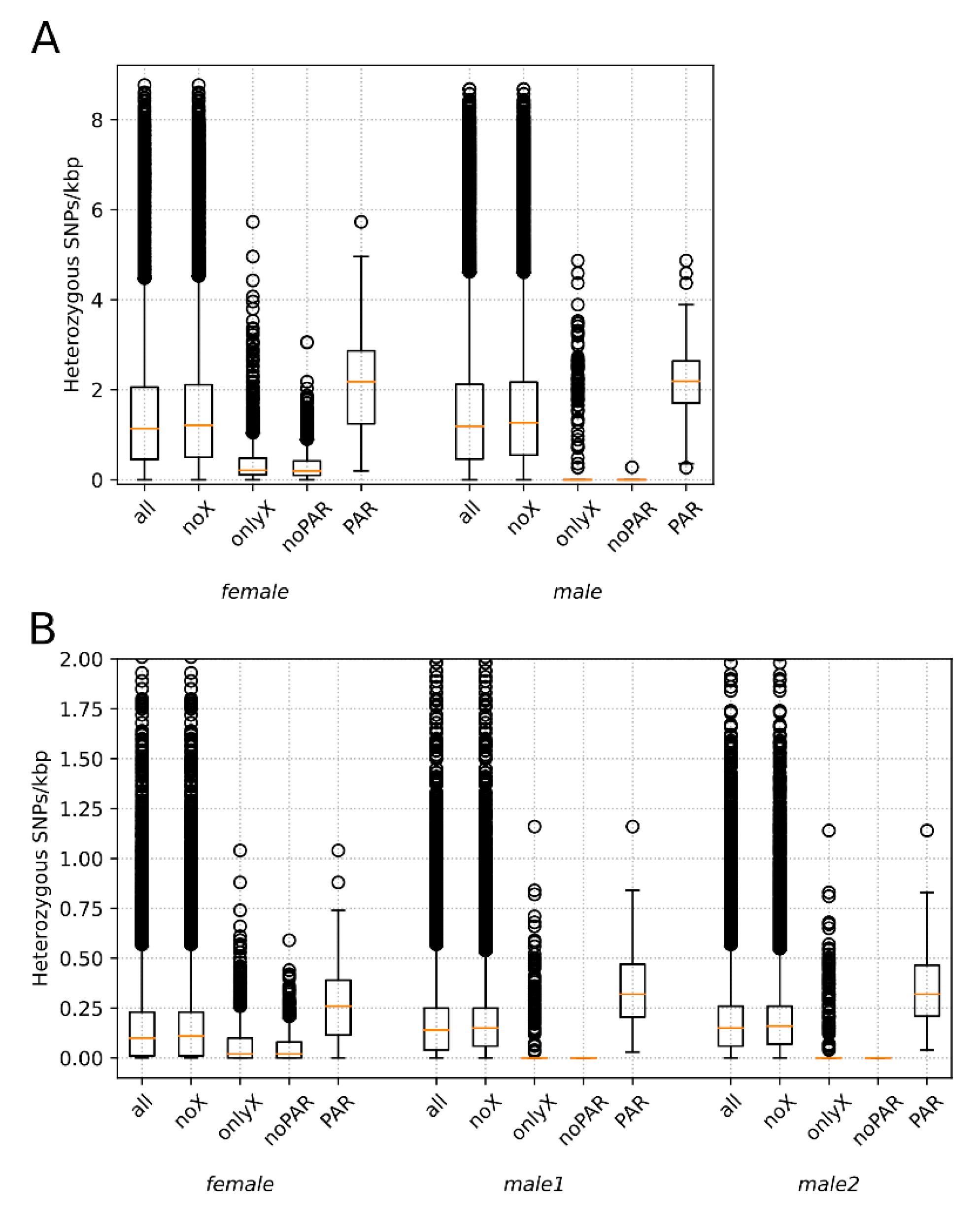
3.2. Heterozygosity Estimations and Visualization
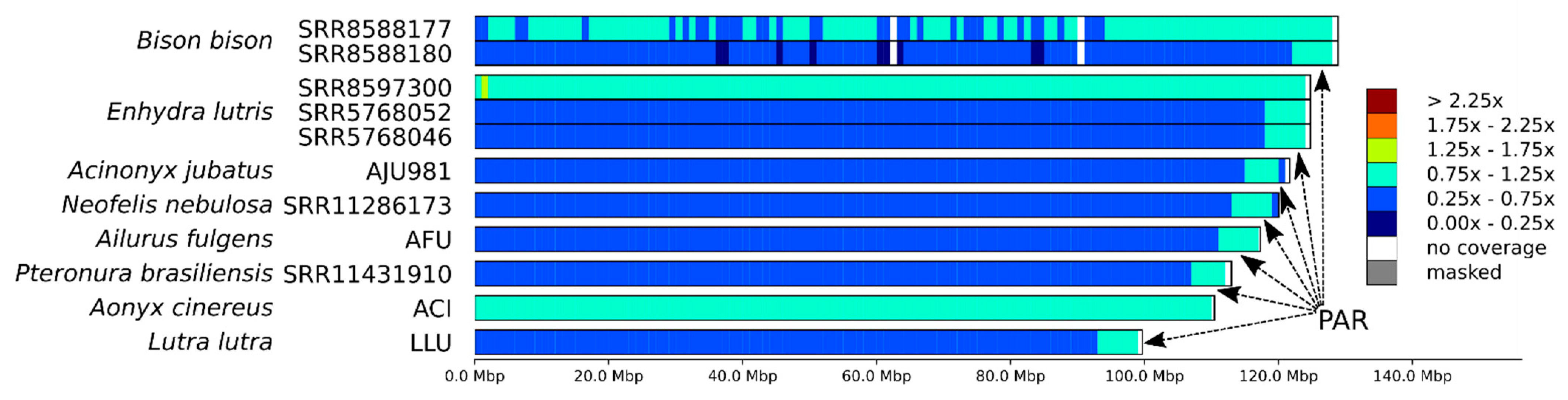
3.3. X Chromosome and the Pseudoautosomal Region
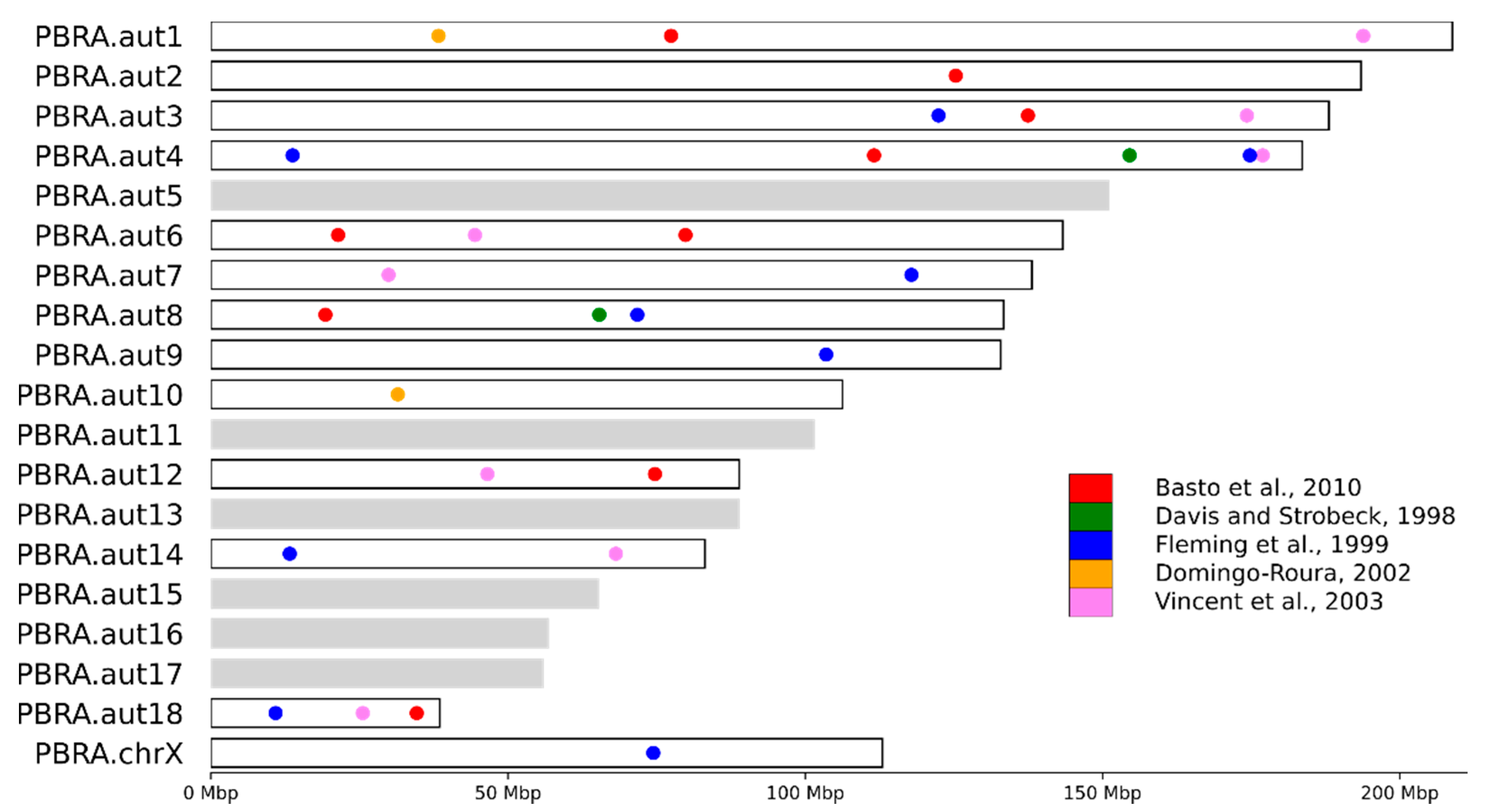
3.4. STR Marker Localization
4. Discussion
4.1. Distribution of Heterozygosity
4.2. Mapping Sex Chromosomes and PAR
4.3. STR Marker Localization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stange, M.; Barrett, R.D.H.; Hendry, A.P. The importance of genomic variation for biodiversity, ecosystems and people. Nat. Rev. Genet. 2021, 22, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Mable, B.K. Conservation of adaptive potential and functional diversity: Integrating old and new approaches. Conserv. Genet. 2019, 20, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Rus Hoelzel, A.; Bruford, M.W.; Fleischer, R.C. Conservation of adaptive potential and functional diversity. Conserv. Genet. 2019, 20, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ellegren, H.; Galtier, N. Determinants of genetic diversity. Nat. Rev. Genet. 2016, 17, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Oleksyk, T.K.; Smith, M.W.; O’Brien, S.J. Genome-wide scans for footprints of natural selection. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 185–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, A.; Sgrò, C. Climate change and evolutionary adaptation. Nature 2011, 470, 479–485. [Google Scholar] [CrossRef]
- Sgrò, C.M.; Lowe, A.J.; Hoffmann, A.A. Building evolutionary resilience for conserving biodiversity under climate change. Evol. Appl. 2011, 4, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Reid, N.M.; Proestou, D.A.; Clark, B.W.; Warren, W.C.; Colbourne, J.K.; Shaw, J.R.; Karchner, S.I.; Hahn, M.E.; Nacci, D.; Oleksiak, M.F.; et al. The genomic landscape of rapid repeated evolutionary adaptation to toxic pollution in wild fish. Science 2016, 354, 1305–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, F.C.; Grabherr, M.G.; Chan, Y.F.; Russell, P.; Mauceli, E.; Johnson, J.; Swofford, R.; Pirun, M.; Zody, M.C.; White, S.; et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 2012, 484, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visser, M.E. Keeping up with a warming world; assessing the rate of adaptation to climate change. Proc. R. Soc. B Biol. Sci. 2008, 275, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visser, M.E. Evolution: Adapting to a Warming World. Current Biology. Curr. Biol. 2019, 18, R1189–R1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.T.; Yeung, C.K.L.; Omland, K.E.; Pang, E.L.; Hao, Y.; Liao, B.Y.; Cao, H.F.; Zhang, B.W.; Yeh, C.F.; Hung, C.M.; et al. Standing genetic variation as the predominant source for adaptation of a songbird. Proc. Natl. Acad. Sci. USA 2019, 116, 2152–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.E.; Koepfli, K. The Role of Genomics in Conservation and Reproductive Sciences. Reprod. Sci. Anim. Conserv. 2014. [Google Scholar] [CrossRef]
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The power and promise of population genomics: From genotyping to genome typing. Nat. Rev. Genet. 2003, 4, 981–994. [Google Scholar] [CrossRef]
- Reed, D.H.; Frankham, R. Correlation between Fitness and Genetic Diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Spielman, D.; Brook, B.W.; Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA 2004, 101, 15261–15264. [Google Scholar] [CrossRef] [Green Version]
- Kirk, H.; Freeland, J.R. Applications and Implications of Neutral versus Non-neutral Markers in Molecular Ecology. Int. J. Mol. Sci. 2011, 12, 3966–3988. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Amos, W.; Balmford, A. When does conservation genetics matter? Heredity 2001, 87, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jost, L.; Archer, F.; Flanagan, S.; Gaggiotti, O.; Hoban, S.; Latch, E. Differentiation measures for conservation genetics. Evol. Appl. 2018, 11, 1139–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, B.J.; Teeling, E.C.; Höglund, J. How and why should we implement genomics into conservation? Evol. Appl. 2014, 7, 999–1007. [Google Scholar] [CrossRef]
- Armstrong, J.; Fiddes, I.T.; Diekhans, M.; Paten, B. Whole-Genome Alignment and Comparative Annotation. Annu Rev Anim Biosci. 2019, 7, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Dudchenko, O.; Shamim, M.S.; Batra, S.S.; Durand, N.C.; Musial, N.T.; Mostofa, R.; Pham, M.; Glenn St Hilaire, B.; Yao, W.; Stamenova, E.; et al. The juicebox assembly tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Wu, Q.; Ma, S.; Ma, T.; Shan, L.; Wang, X.; Nie, Y.; Ning, Z.; Yan, L.; Xiu, Y.; et al. Comparative genomics reveals convergent evolution between the bamboo-eating giant and red pandas. Proc. Natl. Acad. Sci. USA 2017, 114, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Beichman, A.C.; Koepfli, K.P.; Li, G.; Murphy, W.; Dobrynin, P.; Kliver, S.; Tinker, M.T.; Murray, M.J.; Johnson, J.; Lindblad-Toh, K.; et al. Aquatic Adaptation and Depleted Diversity: A Deep Dive into the Genomes of the Sea Otter and Giant Otter. Mol. Biol. Evol. 2019, 36. [Google Scholar] [CrossRef] [PubMed]
- de Manuel, M.; Barnett, R.; Sandoval-Velasco, M.; Yamaguchi, N.; Vieira, F.G.; Lisandra Zepeda Mendoza, M.; Liu, S.; Martin, M.D.; Sinding, M.H.S.; Mak, S.S.T.; et al. The evolutionary history of extinct and living lions. Proc. Natl. Acad. Sci. USA 2020, 117, 10927–10934. [Google Scholar] [CrossRef] [PubMed]
- Dobrynin, P.; Liu, S.; Tamazian, G.; Xiong, Z.; Yurchenko, A.A.; Krasheninnikova, K.; Kliver, S.; Schmidt-Küntzel, A.; Koepfli, K.P.; Johnson, W.; et al. Genomic legacy of the African cheetah, Acinonyx jubatus. Genome Biol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, J.L.; Decker, J.E.; Schnabel, R.D.; Taylor, J.F. Candidate lethal haplotypes and causal mutations in Angus cattle. BMC Genom. 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. Babraham Bioinform. 2020. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 January 2021).
- Kliver, S.F. KrATER (K-Mer Analysis Tool Easy to Run). 2017. Available online: https://github.com/mahajrod/KrATER (accessed on 15 January 2021).
- Starostina, E.; Tamazian, G.; Dobrynin, P.; O’brien, S.; Komissarov, A. Cookiecutter: A tool for kmer-based read filtering and extraction. BioRxiv 2015. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Graphodatsky, A.; Perelman, P.; O’Brien, S.J. Atlas of Mammalian Chromosomes; John Wiley & Sons, Incorporated: Hoboken, NJ, USA, 2020. [Google Scholar]
- Frith, M.C.; Kawaguchi, R. Split-alignment of genomes finds orthologies more accurately. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Basto, M.P.; Rodrigues, M.; Santos-Reis, M.; Bruford, M.W.; Fernandes, C.A. Isolation and characterization of 13 tetranucleotide microsatellite loci in the Stone marten (Martes foina). Conserv. Genet. Resour. 2010, 2, 317–319. [Google Scholar] [CrossRef]
- Davis, C.S.; Strobeck, C. Isolation, variability, and cross-species amplification of polymorphic microsatellite loci in the family mustelidae. Mol. Ecol. 1998, 7, 1776–1778. [Google Scholar] [CrossRef]
- Fleming, M.A.; Ostrander, E.A.; Cook, J.A. Microsatellite markers for american mink (Mustela vison) and ermine (Mustela erminea). Mol. Ecol. 1999, 8, 1352–1355. [Google Scholar] [CrossRef]
- Vincent, I.R.; Farid, A.; Otieno, C.J. Variability of thirteen microsatellite markers in American mink (Mustela vison). Can. J. Anim. Sci. 2003, 83, 597–599. [Google Scholar] [CrossRef] [Green Version]
- Gardner, S.N.; Slezak, T. Simulate_PCR for amplicon prediction and annotation from multiplex, degenerate primers and probes. BMC Bioinform. 2014, 15, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, H.A.; Graves, J.A.M.; Ryder, O.A.; Graphodatsky, A.S.; O’Brien, S.J. Precision nomenclature for the new genomics. GigaScience 2019. [Google Scholar] [CrossRef] [PubMed]
- IUCN. The IUCN Red List of Threatened Species [WWW Document]. Version 2021-1ISSN 2307-8235. 2021. Available online: https://www.iucnredlist.org (accessed on 15 January 2021).
- Domingo-Roura, X. Genetic distinction of marten species by fixation of a microsatellite region. J. Mammal. 2002, 83, 907–912. [Google Scholar] [CrossRef]
- Renaud, G.; Hanghøj, K.; Korneliussen, T.S.; Willerslev, E.; Orlando, L. Joint Estimates of Heterozygosity and Runs of Homozygosity for Modern and Ancient Samples. Genetics 2019, 212, 587–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiblet, W.M.; Zhao, K.; O’Brien, S.J.; Massey, S.E.; Roca, A.L.; Oleksyk, T.K. SmileFinder: A resampling-based approach to evaluate signatures of selection from genome-wide sets of matching allele frequency data in two or more diploid populations. GigaScience 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oleksyk, T.K.; Zhao, K.; de La Vega, F.M.; Gilbert, D.A.; O’Brien, S.J.; Smith, M.W. Identifying selected regions from heterozygosity and divergence using a light-coverage genomic dataset from two human populations. PLoS ONE 2008, 3, e1712. [Google Scholar] [CrossRef] [PubMed]
- Volfovsky, N.; Oleksyk, T.K.; Cruz, K.C.; Truelove, A.L.; Stephens, R.M.; Smith, M.W. Genome and gene alterations by insertions and deletions in the evolution of human and chimpanzee chromosome 22. BMC Genom. 2009, 10. [Google Scholar] [CrossRef] [Green Version]
- Osada, N.; Nakagome, S.; Mano, S.; Kameoka, Y.; Takahashi, I.; Terao, K. Finding the factors of reduced genetic diversity on X chromosomes of Macaca fascicularis: Male-driven evolution, demography, and natural selection. Genetics 2013, 195, 1027–1035. [Google Scholar] [CrossRef] [Green Version]
- Flaquer, A.; Rappold, G.A.; Wienker, T.F.; Fischer, C. The human pseudoautosomal regions: A review for genetic epidemiologists. Eur. J. Hum. Genet. 2008, 16, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, S.P.; Pannell, J.R.; Peichel, C.L.; Ashman, T.-L.; Charlesworth, D.; Chippindale, A.K.; Delph, L.F.; Guerrero, R.F.; Scarpino, S.V.; McAllister, B.F. About PAR: The distinct evolutionary dynamics of the pseudoautosomal region. Trends Genet. 2011, 27, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Sayres, M.A. Genetic Diversity on the Sex Chromosomes. Genome Biol. Evol. 2018, 10, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.J.; Brotman, S.M.; Wilson Sayres, M.A. Genetic Diversity on the Human X Chromosome Does Not Support a Strict Pseudoautosomal Boundary. Genetics 2016, 203, 485–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filatov, D.A.; Gerrard, D.T. High mutation rates in human and ape pseudoautosomal genes. Gene 2003, 317, 67–77. [Google Scholar] [CrossRef]
- Lien, S.; Szyda, J.; Schechinger, B.; Rappold, G.; Arnheim, N. Evidence for heterogeneity in recombination in the human pseudoautosomal region: High resolution analysis by sperm typing and radiation-hybrid mapping. Am. J. Hum. Genet. 2000, 66, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmann, I.; Ebersberger, I.; Ptak, S.E.; Pääbo, S.; Przeworski, M. A neutral explanation for the correlation of diversity with recombination rates in humans. Am. J. Hum. Genet. 2003, 72, 1527–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.W.; Friedman, R.; Yu, N.; Yu, A.; Li, W.H. How strong is the mutagenicity of recombination in mammals? Mol. Biol. Evol. 2005, 22, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.; Ashworth, A. Evolutionary rate of a gene affected by chromosomal position. Curr. Biol. 1999, 9. [Google Scholar] [CrossRef] [Green Version]
- Charlesworth, B. The effects of deleterious mutations on evolution at linked sites. Genetics 2012, 190, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Vicoso, B.; Charlesworth, B. Evolution on the X chromosome: Unusual patterns and processes. Nat. Rev. Genet. 2006, 7, 645–653. [Google Scholar] [CrossRef]
- Zimmerman, S.J.; Aldridge, C.L.; Oyler-McCance, S.J. An Empirical Comparison of Population Genetic Analyses Using Microsatellite and SNP Data for a Species of Conservation Concern. BMC Genom. 2020, 21, 382. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the Power of RADseq for Ecological and Evolutionary Genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, E.; Abramov, A.V.; Soloviev, V.A.; Saveljev, A.P.; Nishita, Y.; Kaneko, Y.; Masuda, R. Hybridization between the European and Asian Badgers (Meles, Carnivora) in the Volga-Kama Region, Revealed by Analyses of Maternally, Paternally and Biparentally Inherited Genes. Mamm. Biol. 2019, 94, 140–148. [Google Scholar] [CrossRef]
- Rozhnov, V.V.; Pishchulina, S.L.; Meschersky, I.G.; Simakin, L.V. On the ratio of phenotype and genotype of sable and pine marten in sympatry zone in the Northern Urals. Mosc. Univ. Biol. Sci. Bull. 2013, 68, 178–181. [Google Scholar] [CrossRef]
- Rhie, A.; McCarthy, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J.; et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature. 2021, 592, 737–746. [Google Scholar] [CrossRef]
- Ceballos, G.; Ehrlich, P.R.; Barnosky, A.D.; García, A.; Pringle, R.M.; Palmer, T.M. Accelerated modern human–induced species losses: Entering the sixth mass extinction. Sci. Adv. 2015, 1, e1400253. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | IUCN Red List Category 1 | Common Name | 2n | Assembly Source or ID | Assembly Level 2 | Length, Gbp | Ns, Mbp | N50, Mbp | dN, % |
|---|---|---|---|---|---|---|---|---|---|
| Aonyx cinereus | VU | Asian small-clawed otter | 38 | DNA Zoo | Chr | 2.44 | 15.5 | 130.94 | +1048% |
| DNA Zoo draft | Draft | 2.42 | 1.35 | 0.1 | |||||
| Enhydra lutris | EN | Sea otter | 38 | DNA Zoo | Chr | 2.45 | 28.94 | 145.94 | −2% |
| GCA_002288905.2 | Draft | 2.46 | 29.68 | 38.75 | |||||
| Lutra lutra | LC | Eurasian river otter | 38 | DNA Zoo | Chr | 2.44 | 0.1 | 148.99 | n/a |
| Pteronura brasiliensis | EN | Giant otter | 38 | DNA Zoo | Chr | 2.46 | 11.89 | 133.38 | +749% |
| DNA Zoo draft | Draft | 2.45 | 1.4 | 0.17 | |||||
| Ailurus fugens | EN | Red panda | 36 | DNA Zoo | Chr | 2.34 | 34.41 | 143.8 | +1% |
| GCA_002007465.1 | Draft | 2.34 | 34.04 | 2.98 | |||||
| Acinonyx jubatus | VU | Cheetah | 38 | DNA Zoo | Chr | 2.37 | 42.86 | 144.64 | +2% |
| GCA_001443585.1 | Draft | 2.37 | 42.06 | 3.12 | |||||
| Neofelis nebulosa | VU | Clouded leopard | 38 | DNA Zoo | Chr | 2.42 | 7.94 | 147.11 | +35% |
| DNA Zoo draft | Draft | 2.41 | 5.89 | 1.38 | |||||
| Bison bison | NT | American bison | 60 | DNA Zoo | Chr | 2.83 | 199.31 | 101.69 | +2% |
| GCF_000754665.1 | Draft | 2.83 | 195.77 | 7.19 |
| Species | STR Markers | #* Chr** | #* Chr** with Markers | #* Chr** w/o Markers | ||
|---|---|---|---|---|---|---|
| Localized (L) | Not Amplified (NA) | Declined (D) | ||||
| Aonyx cinereus1 | 31 | 16 | 19 | 19 | 15 | 4 |
| Enhydra lutris2 | 26 | 22 | 18 | 19 | 14 | 5 |
| Lontra canadensis3 | 28 | 17 | 21 | 19 | 15 | 4 |
| Lutra lutra4 | 26 | 22 | 18 | 19 | 13 | 6 |
| Mustela putorius furo5 | 28 | 17 | 21 | 20 | 14 | 6 |
| Pteronura brasiliensis6 | 30 | 18 | 18 | 19 | 13 | 6 |
| Neovison vison7,*** | 36 | 17 | 13 | 15 | - | - |
| Species | #* Het. SNPs, Millions | Window Size | #*Windows | Median, Het SNPs/kbp | Mean, Het SNPs/kbp | p-Value (Draft vs. Chr.) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Draft | Chr. | Draft | Chr. | Draft | Chr. | Draft | Chr. | Raw | Adjusted | ||
| Aonyx cinereus | 2.73 | 2.73 | 100 kbp | 9777 | 22,183 | 1.100 | 1.190 | 1.052 | 1.144 | 3.37 × 10−34 | 2.36 × 10−33 |
| 1 Mbp | 3 | 2204 | 0.001 | 1.177 | 0.292 | 1.146 | NA | NA | |||
| Enhydra lutris | 0.47 | 0.46 | 100 kbp | 24,146 | 24,165 | 0.140 | 0.140 | 0.178 | 0.182 | 0.98 | 1 |
| 1 Mbp | 2337 | 2396 | 0.174 | 0.176 | 0.175 | 0.180 | 0.79 | 1 | |||
| Pteronura brasilensis | 1.25 | 1.24 | 100 kbp | 13,589 | 22,819 | 0.410 | 0.410 | 0.488 | 0.497 | 0.59 | 1 |
| 1 Mbp | 32 | 2262 | 0.699 | 0.542 | 0.563 | 0.497 | NA | NA | |||
| Ailurus fulgens | 2.14 | 2.14 | 100 kbp | 22,083 | 23,139 | 0.920 | 0.920 | 0.916 | 0.912 | 0.50 | 1 |
| 1 Mbp | 1573 | 2298 | 0.980 | 0.971 | 0.943 | 0.914 | 0.17 | 1 | |||
| Acinonyx jubatus | 0.75 | 0.75 | 100 kbp | 22,861 | 23,609 | 0.280 | 0.280 | 0.314 | 0.314 | 0.42 | 1 |
| 1 Mbp | 1757 | 2350 | 0.332 | 0.330 | 0.322 | 0.314 | 0.23 | 1 | |||
| Neofelis nebulosa | 1.00 | 1.00 | 100 kbp | 22,004 | 23,931 | 0.380 | 0.370 | 0.415 | 0.407 | 6.62 × 10−04 | 0.0046 |
| 1 Mbp | 1194 | 2387 | 0.427 | 0.415 | 0.426 | 0.407 | 1.2 × 10−03 | 0.0089 | |||
| Bison bison | 3.68 | 3.68 | 100 kbp | 24,286 | 26,213 | 1.160 | 1.100 | 1.423 | 1.378 | 5.33 × 10−07 | 3.73 × 10−06 |
| 1 Mbp | 2181 | 2604 | 1.328 | 1.324 | 1.414 | 1.379 | 0.142 | 0.9943 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Totikov, A.; Tomarovsky, A.; Prokopov, D.; Yakupova, A.; Bulyonkova, T.; Derezanin, L.; Rasskazov, D.; Wolfsberger, W.W.; Koepfli, K.-P.; Oleksyk, T.K.; et al. Chromosome-Level Genome Assemblies Expand Capabilities of Genomics for Conservation Biology. Genes 2021, 12, 1336. https://doi.org/10.3390/genes12091336
Totikov A, Tomarovsky A, Prokopov D, Yakupova A, Bulyonkova T, Derezanin L, Rasskazov D, Wolfsberger WW, Koepfli K-P, Oleksyk TK, et al. Chromosome-Level Genome Assemblies Expand Capabilities of Genomics for Conservation Biology. Genes. 2021; 12(9):1336. https://doi.org/10.3390/genes12091336
Chicago/Turabian StyleTotikov, Azamat, Andrey Tomarovsky, Dmitry Prokopov, Aliya Yakupova, Tatiana Bulyonkova, Lorena Derezanin, Dmitry Rasskazov, Walter W. Wolfsberger, Klaus-Peter Koepfli, Taras K. Oleksyk, and et al. 2021. "Chromosome-Level Genome Assemblies Expand Capabilities of Genomics for Conservation Biology" Genes 12, no. 9: 1336. https://doi.org/10.3390/genes12091336
APA StyleTotikov, A., Tomarovsky, A., Prokopov, D., Yakupova, A., Bulyonkova, T., Derezanin, L., Rasskazov, D., Wolfsberger, W. W., Koepfli, K. -P., Oleksyk, T. K., & Kliver, S. (2021). Chromosome-Level Genome Assemblies Expand Capabilities of Genomics for Conservation Biology. Genes, 12(9), 1336. https://doi.org/10.3390/genes12091336