3.1. S. cerevisiae’s Total RNA Composition Is Changed by Chemical Stress Exposure
Intrigued by the concept of stress-dependent RNA modification reprogramming [
4], we set out to study the reaction of
S. cerevisiae on the transcriptome level in more detail. For this purpose, we wanted to use our established NAIL-MS methodology which is based on controlled stable isotope nutrient’s addition to minimal medium [
9]. NAIL-MS relies on yeast nitrogen based medium (YNB), which differs largely from the commonly used yeast extract peptone dextrose medium (YPD). Thus, we first determined the 50% lethal dose of every stressor for a
S. cerevisisae BY4741 culture grown in YNB medium (
Figure S1). For this purpose, an overnight yeast culture was diluted in YNB medium and grown for 3 h until mid-log growth phase and stressed with either methyl-methanesulfonate (MMS) or one of the oxidants: hydrogen peroxide (H
2O
2), arsenite (NaAsO
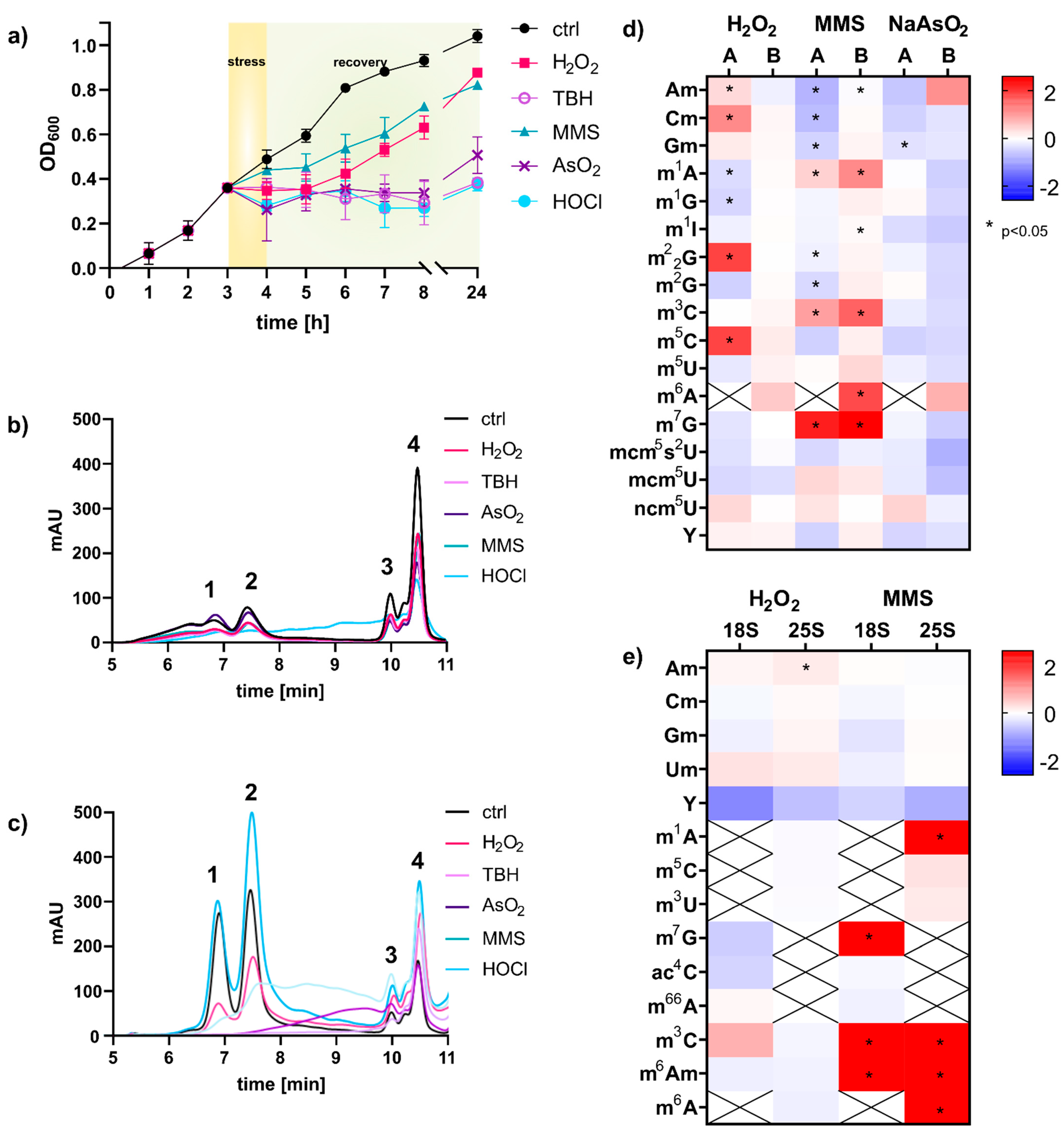
2), tert-butyl hydroperoxide (TBH) or hypochloric acid (HOCl). After one hour of stress exposure, the cells were pelleted and resuspended in fresh YNB medium for recovery. The growth curve for the non-exposed control cells shows the unaltered growth of the cells, whereas the cells exposed to MMS and H
2O
2 showed a delay in growth after stress exposure (
Figure 1). The cells exposed to the oxidants NaAsO
2, TBH and HOCl did not recover within 24 h and showed no growth within this timespan. We next extracted the total RNA from cells after one hour of exposure using either the commercial TRI reagent and glass bead approach or hot phenol [
13]. The total RNA was loaded onto a size exclusion chromatography column of 1000 Å, and the eluting RNA was detected using UV absorption at 254 nm. As shown in
Figure 1b,c, the TRI based method yielded mainly RNAs smaller than 200 nts. 18S and 25S rRNA were of low abundance and undefined size. In contrast, the hot-phenol method yielded high amounts for 18S, 25S and tRNA. Judging from the elution profile in
Figure 1c, the integrity of rRNAs remained under MMS and H
2O
2 exposure, whereas all rRNA was lost in cells exposed to the oxidants NaAsO
2, TBH and HOCl. Overall, the profile of total RNA was bizarre in these cells, and the fate of the rRNA is unclear. Therefore, the stressor HOCl was not further pursued for RNA modification analysis. Only NaAsO
2 and TBH showed acceptable integrity of tRNA, and thus tRNA modification profiles can be analyzed.
3.2. tRNA Modification Reprogramming in S. cerevisiae Is Stress Dependent; rRNA Modifications Are Unaltered
For RNA modification analysis we used our established stable isotope dilution LC-MS/MS protocol [
12]. In the acute phase, 60 min after stress exposure, we found changes in tRNA modification density in dependence of the chemical used, as previously suggested by Chan et al. [
4]. A comparison of the published data and our fold-change data is given in
Figure 1d. The direct comparison revealed several differences between our and the published data. For H
2O
2, we found less tRNA modification reprogramming, while similar trends are found for MMS and NaAsO
2 exposure. We have identified three major experimental differences which contributed to the observed differences: (1) The published experiments were performed in rich YPD growth medium, whereas we used minimal YNB medium. (2) Chan et al. used a column affinity-based protocol for purification of RNA smaller than 200 nts, whereas we used size exclusion chromatography for tRNA purification. (3) Our mass spectrometric data was acquired using stable isotope dilution, which allows absolute quantification of RNA modifications. Therefore, we are confident that our data reflect the changes in tRNA modification profiles accurately. With our study we confirm the findings by Chan et al. that tRNA modifications are reprogrammed in the acute moment of chemical stress exposure and that the changes are dependent on the chemical stressor. However, especially for H
2O
2 exposure, the observed changes were minimal and were not statistically significant. For MMS, we found substantial formation of 1-methyladenosine (m
1A), 3-methylcytidine (m
3C), 6-methyladenosine (m
6A) and 7-methylguanosine (m
7G), as recently described as RNA main damage products [
10,
15]. From the same experiments, we purified the 18S and 25S rRNA and subjected them to RNA modification quantification by LC-MS/MS. After 60 min of stress exposure, we found only minor changes in the natural epitranscriptomes of both rRNAs. This is in accordance with a recent study from the Novoa laboratory [
5]. However, in rRNA from MMS exposed yeast, we found high numbers of the potential damage products m
1A, m
7G, m
3C and m
6A and a damage-methylated 2′-O-methyladenosine (m
xAm).
3.3. MMS Directly Methylates tRNA and rRNA in S. cerevisiae
After MMS exposure, we detected high abundances of those RNA modifications, which are RNA damage products that were described in
E. coli studies [
10,
15]. With the goal of elucidating the origins of these RNA modifications in
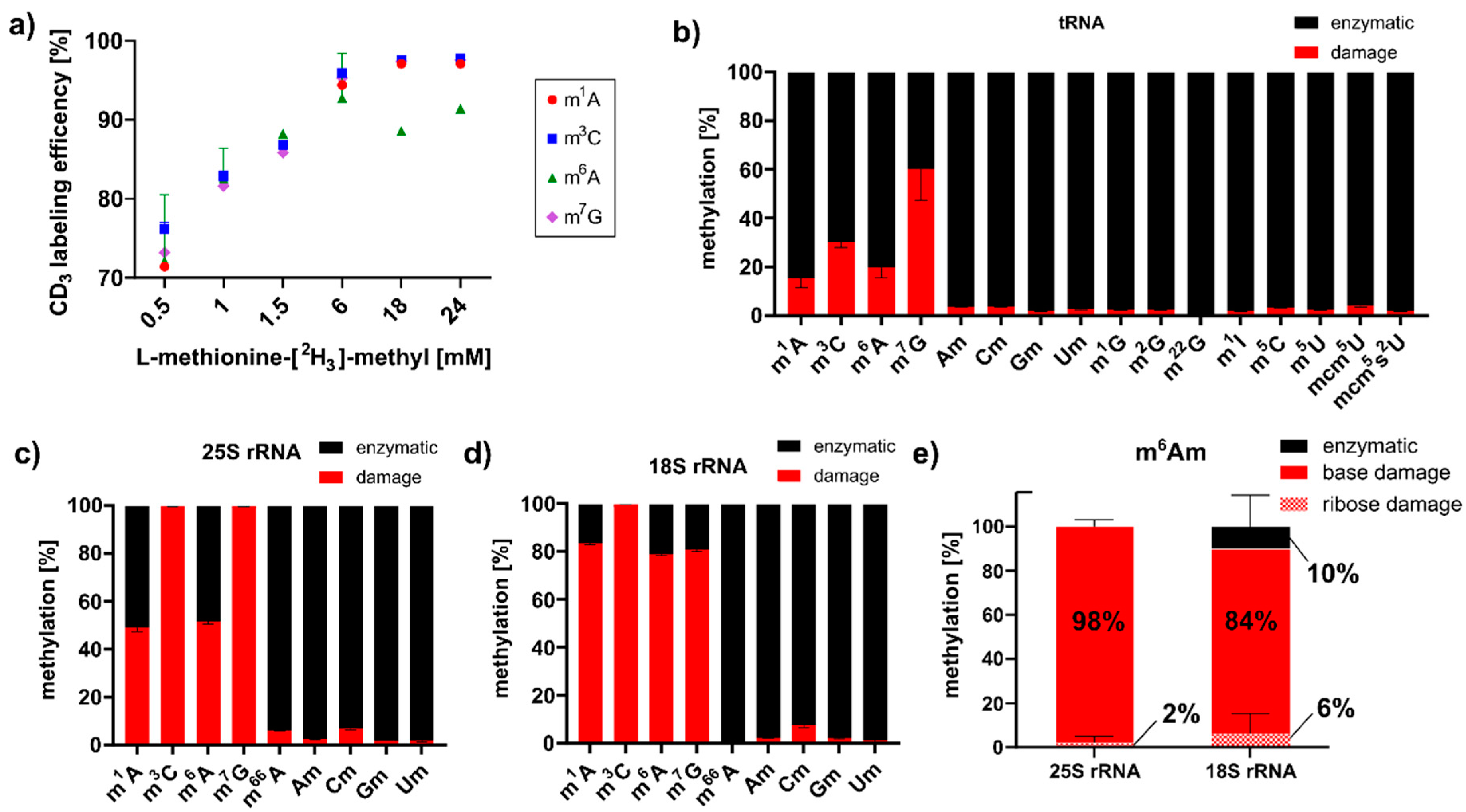
S. cerevisiae, we envisioned a methylation discrimination assay which distinguishes enzymatic RNA methylation from direct methylation damage. S-Adenosylmethionine (SAM) is the natural methyl-donor for yeast RNA methyltransferases, and by feeding L-methionine-[
2H
3]-methyl enzymatic methylations, they receive a +3 mass increase. As shown in
Figure 2a, optimal labeling of native RNA modifications m
7G, m
1A and m
3C was achieved with 18 mM L-methionine-[
2H
3]-methyl. Cells were exposed to 12 mM MMS in the continuous presence of 18 mM L-methionine-[
2H
3]-methyl. After one hour of MMS exposure, tRNA, 18S and 25S rRNA were extracted, and mass spectrometry analysis revealed the ratio of enzymatically placed methylations (
m/
z +3) to damage-derived methylations (
m/
z ± 0). As shown in
Figure 2b, up to 60% of all m
7G marks were caused by direct methylation with MMS. To a lower extent, m
3C, m
1A and m
6A were caused by direct methylation of canonical nucleosides in tRNA. For rRNA, we found the same damage products (
Figure 2c,d), and in addition, two base-methylated 2′-O-methyladenosine species designated as m
xAm. A comparison to our synthetic standards of m
1Am and m
6Am indicates that the early eluting damage product was m
1Am and the later one was m
6Am (
Figure S2). Both were caused by direct base methylation of the highly abundant Am of both rRNAs during MMS exposure. With the power of our methylome discrimination assay, we could clearly identify the origin of the base methylation from MMS and the enzymatic origin of the ribose methylation (
Figure 2e). Intrigued by this finding, we searched for a methylation damage product of Gm in rRNA, and we observed a clear signal of m
7Gm in the MMS exposed yeast samples. Further identification through the comparison with a synthetic standard has not yet been possible (
Figure S2). A detailed analysis of the observed absolute quantities is given
Table S2.
3.4. Modification Density in Existing tRNAs Rises upon S. cerevisiae Stress Exposure
Although the changes in tRNA modification density were small, we were curious to find out how they emerged mechanistically. With a pulse chase NAIL-MS experiment, we aimed to answer the questions: How does transcription change due to stress exposure? Are existing tRNAs degraded? Is it the original tRNAs which are modified or even demodified? When do new transcripts emerge, and how quickly are they modified? To answer these questions, we required a robust NAIL-MS method which allows accurate and precise analysis of mainly methylated nucleosides. In our previous method published in 2017 [
9], we established the necessary medium for such an experiment; however, the stable isotope labeled internal standard (SILIS) was problematic. For methylated cytidine derivatives especially, the same
m/
z was found in the SILIS, along with the original modification of the tRNA. Therefore, a new SILIS, without
m/
z overlap, with the analytes had to be produced. For this purpose, we utilized a commercially available yeast medium which was enriched with carbon-13 and nitrogen-15 instead of carbon-12 and nitrogen-14 atoms. For
S. cerevisiae, glucose is the carbon source and an ideal energy source, and by addition of 0.1 g/L of
13C
6-glucose, we received well growing cultures and high numbers of fully
13C-labeled yeast cells. The total
13C- and
15N-labeled RNA was isolated, the rRNA and tRNA were purified and the SILIS was prepared following our established protocol [
12]. A comparison of our new SILIS and the previous SILIS is found in
Figure S4 and our recently published protocols [
14].
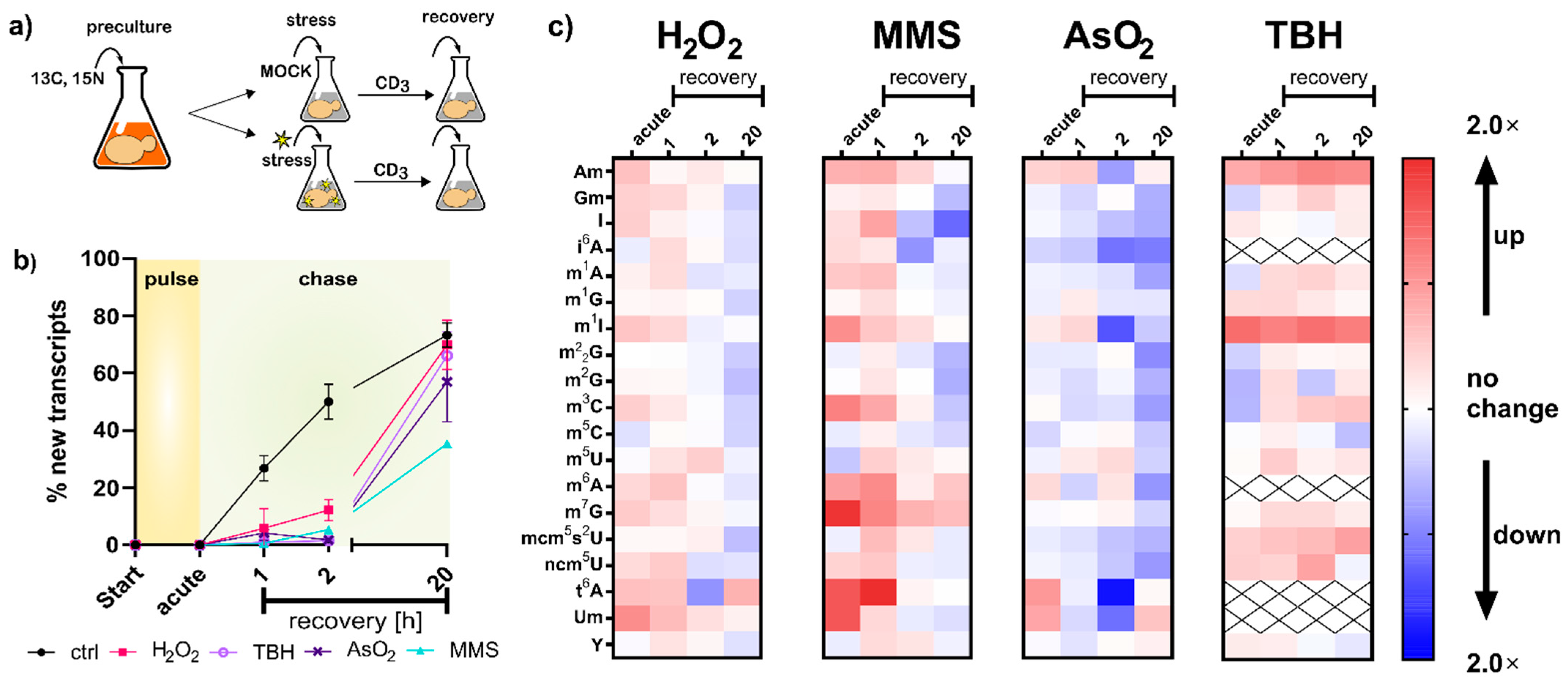
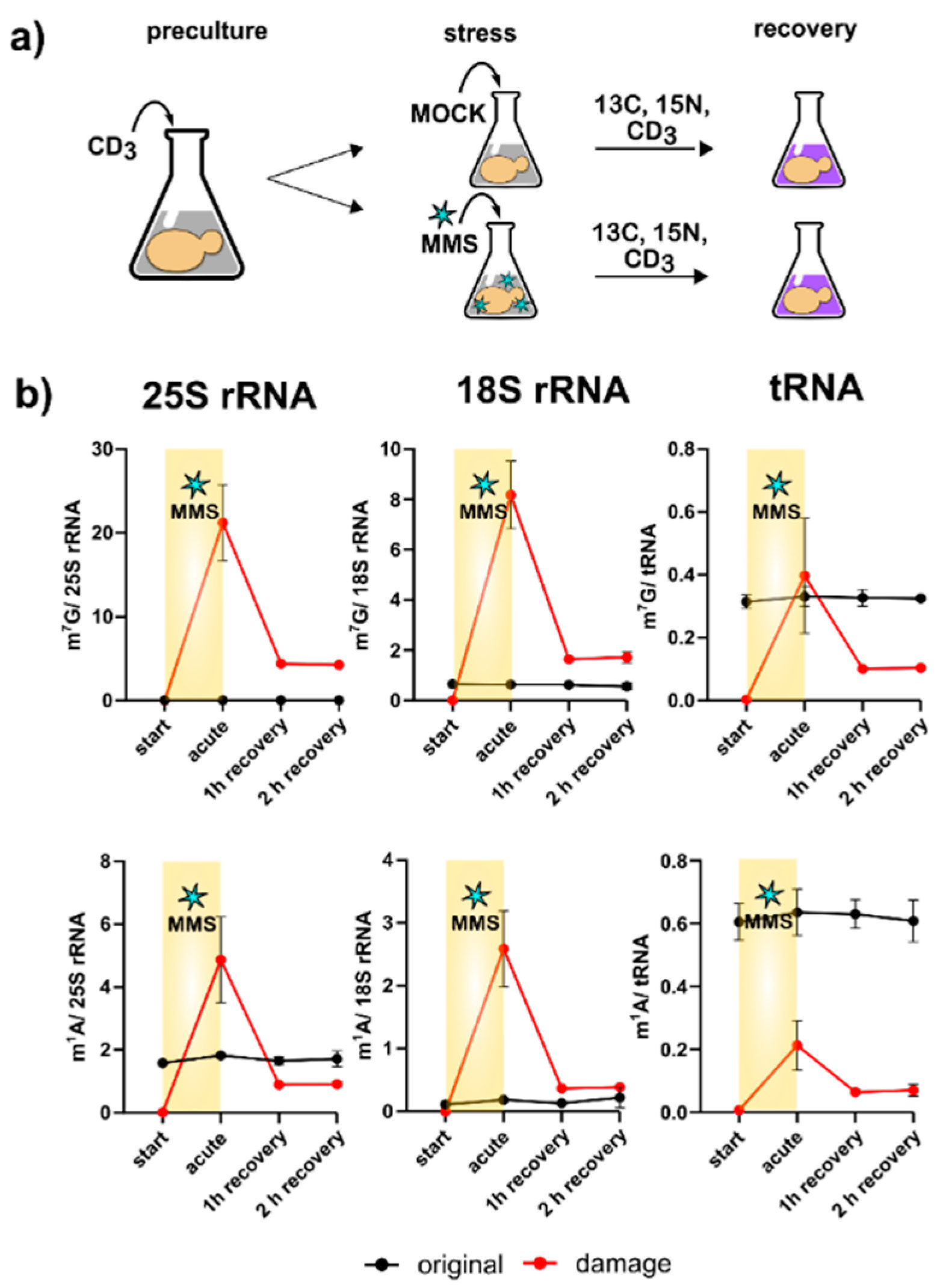
With the new SILIS in hand, we followed our published yeast NAIL-MS protocol (
Figure 3a). Briefly summarized, yeast is grown overnight in YNB medium supplemented with
15N
2-uracil and
13C
6-glucose. The next day, cells were brought to OD 1 in the same medium, left for 3 h to enter mid-log phase and then exposed to the chemical stressor. After one hour of exposure, the stressor was removed by medium exchange. For the chase phase, medium with L-methionine-[
2H
3]-methyl was used. Due to the mass spectrometric detection, we followed the abundances of modified nucleosides in RNA existing during exposure to the chemical, determined the abundance of new canonical nucleosides forming after stress and determined the modification incoperation in new transcripts.
In the first step, we compared the number of new canonical nucleosides to the number of original canonical nucleosides, which is an indicator of cellular metabolism. After two hours of growth in the new but stable isotope labeled medium, 50% of all tRNAs contained new canonical nucleosides. In contrast, all stressed cells contained less than 10% of new canonical nucleosides, which indicates that transcription of tRNA is substantially repressed during the stress recovery phase (
Figure 3b). Thus, all RNA modification density changes observed in
Figure 1d must have been derived from from changed modification patterns in the original tRNAs. To further investigate the impacts of stress on the tRNA modification profiles, we determined the number of modifications per original tRNA and compared the quantities of stressed cells to the numbers found in the respective control cells. Under acute stress, we mainly observed higher numbers of tRNA modifications for H
2O
2, MMS and TBH stress and lower numbers for NaAsO
2 stress (
Figure 3c). These observations are in good agreement with our findings in
Figure 1d.
Regarding the overarching hypothesis of stress-dependent RNA modification reprogramming, our findings concerning transcription rates (
Figure 3b) indicate a non-active adaptation scenario for increases in tRNA modification abundance. The increase in tRNA modification density might have been caused by a combination of (a) halted transcription in stressed cells, which left only existing transcripts as substrates for RNA writer enzymes, and thus higher modification numbers were observed; and (b) ongoing transcription and slow maturation in the control cells, and thus we found lower modification numbers in the control cells. Lower numbers of modifications, as observed for NaAsO
2, might indicate active removal of modifications; however, given the large number of different modifications that are affected, a global effect on mature tRNA might be causative. In theory, mature and thus modified tRNAs might be targeted for degradation. Yet, we see no evidence of increased degradation of original tRNAs (
Figure 3b). The trend of decreased tRNA modification density in arsenite-exposed
S. cerevisiae remained visible even after two hours of recovery. Our methodology is currently not able to determine the biological mechanism behind this intriguing observation. A more detailed overview and absolute numbers of all native modifications per tRNA, 18S and 25S rRNA can be found in
Figure S8.
One of the most striking findings of
Figure 3c is the direct methylation of nucleobases, which appears to have been reverted during the recovery phase. We designed a pulse chase assay based on the methylome discrimination methodology to follow the fate of enzymatically placed modified nucleosides and damage-derived nucleosides (
Figure 4a). On average, both rRNAs receive more than a dozen damage methylations during the experiment, and at least one methylation damage is found per tRNA. We found for both tRNA and rRNA, unexpectedly fast loss of these damages within one hour of recovery. The abundance of native methylations remained unchanged (
Figure 4b and
Figure S6). We tested several knockout strains of known nucleic acid damage repair enzymes for their potential involvement in the demethylation process in
S. cerevisiae. However, except for met18, which showed a decent involvement in total RNA demethylation, no enzyme was found to be part of an active demethylation machinery (
Figure S7 and
Table S4). Loss of damaged RNAs through targeted degradation of the damaged RNA subpopulation is another valid hypothesis, but our data on original-to-new transcript ratios in
Figure 3B do not favor this hypothesis. In summary, rRNA and tRNA receive substantial methylation damage through MMS exposure of
S. cerevisiae, but we do not know by which mechanism the damaged nucleosides were lost within 60 min of recovery.
3.5. tRNA Modification Placement in New Transcripts Is Stress Dependent
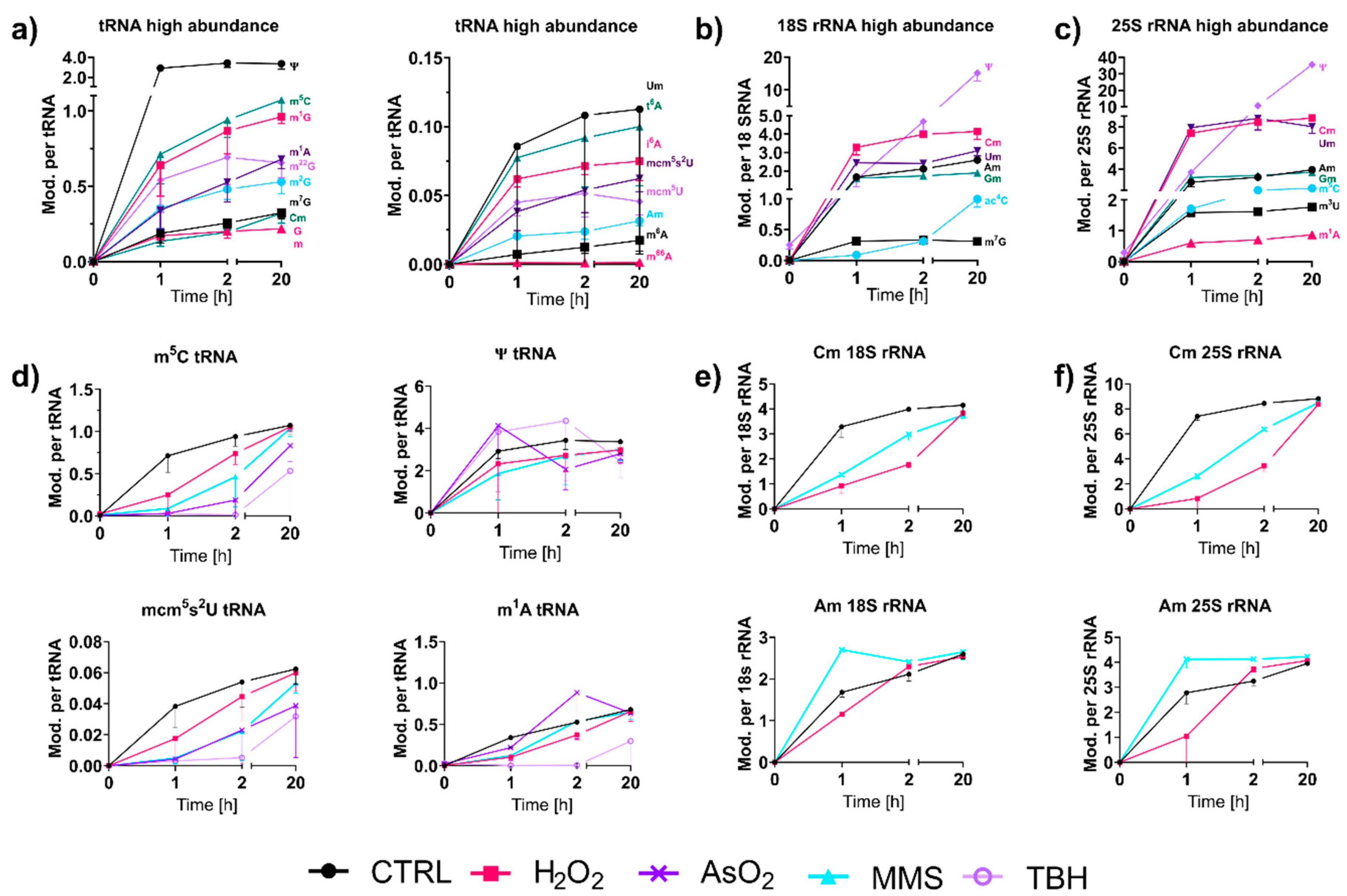
With NAIL-MS, we have the unique opportunity to observe the speed of modifications in RNAs transcribed after a stress event.
Figure 5a shows the early formation of highly abundant and less abundant modifications in total tRNA from
S. cerevisiae. In accordance with our results from 2017 [
9], we found immediately high amounts for modified nucleosides, such as pseudouridine (Ψ) and 2′-O-methylguanosine (Gm). Interestingly, other modifications of the anticodon-stem-loop mcm
5(s
2)U, t
6A and i
6A also appeared early on in total tRNA (
Figure 5d and
Figure S9). For Ψ, an immediate placement was also observed in human total tRNA, human tRNA
Phe [
11] and yeast tRNA
Phe [
16]. However, for Gm, which is incorporated fast in yeast tRNA, we found slow incorporation in human tRNA. Other modified nucleosides such as m
1A, m
5C, Cm and m
7G showed similar incorporation speeds to human tRNAs [
11].
For ribosomal RNAs, we found an immediate steady-state abundance of ribose methylated modifications, as expected (
Figure 5b) [
17]. In 18S rRNA, m
7G and in 25S rRNA, m
3U, m
5C and m
1A, were also placed early on, which is unsurprising, given their later inaccessibility to modification enzymes in the mature ribosome. Previous studies proposed rRNA methylation as a co-transcriptional process in the early phase of ribosome assembly [
17,
18]. To our great surprise, the isomerization of uridine to Ψ was substantially slower, and it took more than two hours to reach the final modification density in both 18S and 25S rRNA. This observation is in stark contrast to our findings in human rRNA, where we observed a fast pseudouridinylation within minutes [
11]. Our most puzzling result was observed for Ψ in the latest 20 h timepoint. Its abundance exceeded the steady state level observed in the unlabeled experiment (
Table S2). We excluded a methodological bias, due to the biologically valid data received for tRNA. Thus, this observation deserves continued research to discover the underlying mechanism.
For most tRNA and rRNA modifications, we observed immediate or extremely fast incorporation within one hour of transcription (
Figure 5a–c). From our NAIL-MS stress exposure studies in
Figure 3b, we now know that transcription is differentially impacted by the chosen stressors. However, how about the subsequent RNA modification processes? To address this yet unsolved question, we analyzed the emergence of modified nucleosides within new transcripts in the recovery phase using the NAIL-MS experiment from
Figure 3c. For new tRNAs, we observed delayed incorporation of most modified nucleosides. This was exemplarily shown for m
5C and mcm
5s
2U in
Figure 5d, but also for Cm, m
3C, Gm, m
1G, m
2G, m
22G and m
7G. For most modified nucleosides, the incorporation delay was strongest for the oxidants TBH and NaAsO
2, and H
2O
2 and MMS had more modest delays. For rRNA, a similar delay in modification speed was observed. Cm formation was especially slower under stress in comparison to unstressed cells (
Figure 5e,f). In contrast, m
1A and Ψ in tRNA and Am in both rRNAs were incorporated as fast or even faster after stress and to a higher degree compared to the unstressed controls. In yeast tRNA
Phe, m
1A and Ψ were recently shown to be the starting point during maturation, which indicates their important role in this tRNA’s modification network [
16]. NaAsO
2 appeared to have increased m
1A and Ψ abundances in newly transcribed RNAs, which might indicate involvement of these modified nucleosides in the arsenite stress response.
Oxidative stress has a substantial impact on the cell, and thiolated biomolecules suffer especially from exposure to reactive oxidant species. For example, bacterial DNA can be naturally thiolated at a non-bridging oxygen of the phosphodiester bond. During exposure to hypochloric acid, the sulfur can be replaced by oxygen, which either causes lethal strand breaks or a regular phosphodiester bond [
19]. Our NAIL-MS data from NaAsO
2 and TBH stressed cells showed a potentially similar effect on thiolated tRNA modifications. In eukaryotes, methylthiolation of adenine at position 2 has been reported, and the resulting modifications ms
2i
6A and ms
2t
6A are known to reside in the anticodon stem-loop of tRNAs [
3]. As shown in
Figure S9A, the abundance of original t
6A increased during TBH exposure. In addition, both i
6A and t
6A were substantially more abundant in new tRNA transcripts compared to tRNAs from unstressed controls (
Figure S9). Our NAIL-MS data indicate that thiolated nucleosides might be either direct substrates to reactive oxygen species or are impacted by the disturbed sulfur homeostasis.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




