
Morning Cortisol and Circulating Inflammatory Cytokine Levels: A Mendelian Randomisation Study

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Overview
2.2. Data Sources
2.3. Statistical Analysis
3. Results
4. Discussion
4.1. Principal Findings in Context
4.2. Clinical Implications
4.3. Strengths
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Russell, G.; Lightman, S. The human stress response. Nat. Rev. Endocrinol. 2019, 15, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busillo, J.M.; Cidlowski, J.A. The five Rs of glucocorticoid action during inflammation: Ready, reinforce, repress, resolve, and restore. Trends Endocrinol. Metab. 2013, 24, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Kristofic, C. Regulation by corticosteroids of Th1 and Th2 cytokine production in human CD4+ effector T cells generated from CD45RO- and CD45RO+ subsets. J. Immunol. 1995, 155, 3322–3328. [Google Scholar] [PubMed]
- Wiegers, G.J.; Reul, J.M. Induction of cytokine receptors by glucocorticoids: Functional and pathological significance. Trends Pharmacol. Sci. 1998, 19, 317–321. [Google Scholar] [CrossRef]
- Rahman, R.P.; McEwan, L.; Ryan, D.K.; Gill, D. Leveraging genetic data to investigate the effects of interleukin-6 receptor signalling on levels of 40 circulating cytokines. Br. J. Clin. Pharmacol. 2021; Epub ahead of print. [Google Scholar] [CrossRef]
- Smith, G.D.; Ebrahim, S. “Mendelian randomization”: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emdin, C.A.; Khera, A.V.; Kathiresan, S. Mendelian randomization. JAMA 2017, 318, 1925–1926. [Google Scholar] [CrossRef]
- Davies, N.M.; Holmes, M.V.; Davey Smith, G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 2018, 362, 601. [Google Scholar] [CrossRef] [Green Version]
- Kalaoja, M.; Corbin, L.J.; Tan, V.Y.; Ahola-Olli, A.V.; Havulinna, A.S.; Santalahti, K.; Pitkänen, N.; Lehtimäki, T.; Lyytikäinen, L.P.; Raitoharju, E.; et al. The Role of Inflammatory Cytokines as Intermediates in the Pathway from Increased Adiposity to Disease. Obesity 2021, 29, 428–437. [Google Scholar] [CrossRef]
- Available online: http://www.nealelab.is/uk-biobank/ (accessed on 15 December 2021).
- Burgess, S.; Davies, N.M.; Thompson, S.G. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 2016, 40, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Timpson, N.J.; Higgins, J.P.T.; Dimou, N.; Langenberg, C.; et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomisation (STROBE-MR): Explanation and elaboration. BMJ 2021, 375, n2233. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.A.; Bankier, S.; Altmaier, E.; Barnes, C.L.; Clark, D.W.; Ermel, R.; Friedrich, N.; van der Harst, P.; Joshi, P.K.; Karhunen, V.; et al. Variation in the SERPINA6/SERPINA1 locus alters morning plasma cortisol, hepatic corticosteroid binding globulin expression, gene expression in peripheral tissues, and risk of cardiovascular disease. J. Hum. Genet. 2021, 66, 625–636. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Bolton, J.L.; Hayward, C.; Direk, N.; Lewis, J.G.; Hammond, G.L.; Hill, L.A.; Anderson, A.; Huffman, J.; Wilson, J.F.; Campbell, H.; et al. Genome wide association identifies common variants at the SERPINA6/SERPINA1 locus influencing plasma cortisol and corticosteroid binding globulin. PLoS Genet. 2014, 10, e100447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahola-Olli, A.V.; Würtz, P.; Havulinna, A.S.; Aalto, K.; Pitkänen, N.; Lehtimäki, T.; Kähönen, M.; Lyytikäinen, L.-P.; Raitoharju, E.; Seppälä, I.; et al. Genome-wide Association Study Identifies 27 Loci Influencing Concentrations of Circulating Cytokines and Growth Factors. Am. J. Hum. Genet. 2017, 100, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MRBase platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Small, D.S.; Thompson, S.G. A review of instrumental variable estimators for Mendelian randomization. Stat. Methods Med. Res. 2017, 26, 2333–2355. [Google Scholar] [CrossRef] [Green Version]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.; Georgakis, M.K.; Walker, V.M.; Schmidt, A.F.; Gkatzionis, A.; Freitag, D.F.; Finan, C.; Hingorani, A.D.; Howson, J.M.M.; Burgess, S.; et al. Mendelian randomization for studying the effects of perturbing drug targets. Wellcome Open Res. 2021, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M.; Walz, A.; Kunkel, S.L. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Investig. 1989, 84, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Roger, T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat. Rev. Immunol. 2003, 3, 791–800. [Google Scholar] [CrossRef]
- Roger, T.; David, J.; Glauser, M.P.; Calandra, T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 2001, 414, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Roger, T.; Froidevaux, C.; Martin, C.; Calandra, T. Macrophage migration inhibitory factor (MIF) regulates host responses to endotoxin through modulation of Toll-like receptor 4 (TLR4). J. Endotoxin. Res. 2003, 9, 119–123. [Google Scholar] [CrossRef]
- Koebernick, H.; Grode, L.; David, J.R.; Rohde, W.; Rolph, M.S.; Mittrücker, H.W.; Kaufmann, S.H. Macrophage migration inhibitory factor (MIF) plays a pivotal role in immunity against Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 2002, 99, 13681–13686. [Google Scholar] [CrossRef] [Green Version]
- Mukaida, N.; Gussella, G.L.; Kasahara, T.; Ko, Y.; Zachariae, C.O.; Kawai, T.; Matsushima, K. Molecular analysis of the inhibition of interleukin-8 production by dexamethasone in a human fibrosarcoma cell line. Immunology 1992, 75, 674–679. [Google Scholar]
- Calandra, T.; Bernhagen, J.; Metz, C.N.; Spiegel, L.A.; Bacher, M.; Donnelly, T.; Cerami, A.; Bucala, R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature 1995, 377, 68–71. [Google Scholar] [CrossRef]
- Leech, M.; Metz, C.; Hall, P.; Hutchinson, P.; Gianis, K.; Smith, M.; Weedon, H.; Holdsworth, S.R.; Bucala, R.; Morand, E.F. Macrophage migration inhibitory factor in rheumatoid arthritis: Evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999, 42, 1601–1608. [Google Scholar] [CrossRef]
- Bacher, M.; Metz, C.N.; Calandra, T.; Mayer, K.; Chesney, J.; Lohoff, M.; Gemsa, D.; Donnelly, T.; Bucala, R. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc. Natl. Acad. Sci. USA 1996, 93, 7849–7854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, M.C.; Elsbury, S.K.; Pavli, P.; Doe, W.F. Interleukin 8: Cells of origin in inflammatory bowel disease. Gut 1996, 38, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izutani, R.; Loh, E.Y.; Reinecker, H.C.; Ohno, Y.; Fusunyan, R.D.; Lichtenstein, G.R.; Rombeau, J.L.; Macdermott, R.P. Increased expression of interleukin-8 mRNA in ulcerative colitis and Crohn’s disease mucosa and epithelial cells. Inflamm. Bowel. Dis. 1995, 1, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, L.; Hauser, C.; Zgraggen, K.; Wagner, H.; Hess, M.; Laissue, J.A.; Mueller, C. Expression of interleukin-8 gene in inflammatory bowel disease is related to the histological grade of active inflammation. Am. J. Pathol. 1994, 144, 997–1007. [Google Scholar]
- Arnott, I.D.; Drummond, H.E.; Ghosh, S. Gut Mucosal Secretion of Interleukin 1β and Interleukin-8 Predicts Relapse in Clinically Inactive Crohn’s Disease. Dig. Dis. Sci. 2001, 46, 402–409. [Google Scholar] [CrossRef]
- Oliver, J.; Márquez, A.; Gómez-Garcia, M.; Martinez, A.; Mendoza, J.L.; Vilchez, J.R.; López-Nevot, M.A.; Piñero, A.; de la Concha, E.G.; Nieto, A.; et al. Association of the macrophage migration inhibitory factor gene polymorphisms with inflammatory bowel disease. Gut 2007, 56, 150–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, Y.; Hisamatsu, T.; Kamada, N.; Kitazume, M.T.; Honda, H.; Oshima, Y.; Saito, R.; Takayama, T.; Kobayashi, T.; Chinen, H.; et al. Monocyte chemoattractant protein-1 contributes to gut homeostasis and intestinal inflammation by composition of IL-10-producing regulatory macrophage subset. J. Immunol. 2010, 184, 2671–2676. [Google Scholar] [CrossRef] [Green Version]
- Georganas, C.; Liu, H.; Perlman, H.; Hoffmann, A.; Thimmapaya, B.; Pope, R.M. Regulation of IL-6 and IL-8 expression in rheumatoid arthritis synovial fibroblasts: The dominant role for NF-kappa B but not C/EBP beta or c-Jun. J. Immunol. 2000, 165, 7199–7206. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.Y.; Kim, J.Y.; Kim, K.W.; Park, M.K.; Moon, Y.; Kim, W.U.; Kim, H.Y. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-κB- and PI3-kinase/Akt-dependent pathways. Arthritis Res. Ther. 2004, 6, R120. [Google Scholar] [CrossRef] [Green Version]
- Morand, E.F.; Leech, M.; Weedon, H.; Metz, C.; Bucala, R.; Smith, M.D. Macrophage migration inhibitory factor in rheumatoid arthritis: Clinical correlations. Rheumatology 2002, 41, 558–562. [Google Scholar] [CrossRef] [Green Version]
- Baugh, J.A.; Chitnis, S.; Donnelly, S.C.; Monteiro, J.; Lin, X.; Plant, B.J.; Wolfe, F.; Gregersen, P.K.; Bucala, R. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002, 3, 170–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoscoy-Ascencio, G.; Baños-Hernández, C.J.; Navarro-Zarza, J.E.; Hernández-Bello, J.; Bucala, R.; López-Quintero, A.; Valdés-Alvarado, E.; Parra-Rojas, I.; Illades-Aguiar, B.; Muñoz-Valle, J.F. Macrophage migration inhibitory factor promoter polymorphisms are associated with disease activity in rheumatoid arthritis patients from Southern Mexico. Mol. Genet. Genomic. Med. 2020, 8, e1037. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.M.; Zhao, C.N.; Liu, L.N.; Wu, Q.; Dan, Y.L.; Wang, D.G.; Pan, H.F. Increased circulating interleukin-8 levels in systemic lupus erythematosus patients: A meta-analysis. Biomark. Med. 2018, 12, 1291–1302. [Google Scholar] [CrossRef]
- Ruchakorn, N.; Ngamjanyaporn, P.; Suangtamai, T.; Kafaksom, T.; Polpanumas, C.; Petpisit, V.; Pisitkun, T.; Pisitkun, P. Performance of cytokine models in predicting SLE activity. Arthritis Res. Ther. 2019, 21, 287. [Google Scholar] [CrossRef] [Green Version]
- Rovin, B.H.; Lu, L.; Zhang, X. A novel interleukin-8 polymorphism is associated with severe systemic lupus erythematosus nephritis. Kidney Int. 2002, 62, 261–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, T.; Foote, A.; Lee, J.P.; Morand, E.F.; Harris, J. MIF: Implications in the Pathoetiology of Systemic Lupus Erythematosus. Front. Immunol. 2015, 6, 577. [Google Scholar] [CrossRef] [Green Version]
- Foote, A.; Briganti, E.M.; Kipen, Y.; Santos, L.; Leech, M.; Morand, E.F. Macrophage migration inhibitory factor in systemic lupus erythematosus. J. Rheumatol. 2004, 31, 268–273. [Google Scholar]
- Lan, H.Y.; Yang, N.; Nikolic-Paterson, D.J.; Yu, X.Q.; Mu, W.; Isbel, N.M.; Metz, C.N.; Bucala, R.; Atkins, R.C. Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int. 2000, 57, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Hoi, A.Y.; Iskander, M.N.; Morand, E.F. Macrophage migration inhibitory factor: A therapeutic target across inflammatory diseases. Inflamm. Allergy Drug Targets 2007, 6, 183–190. [Google Scholar] [CrossRef]



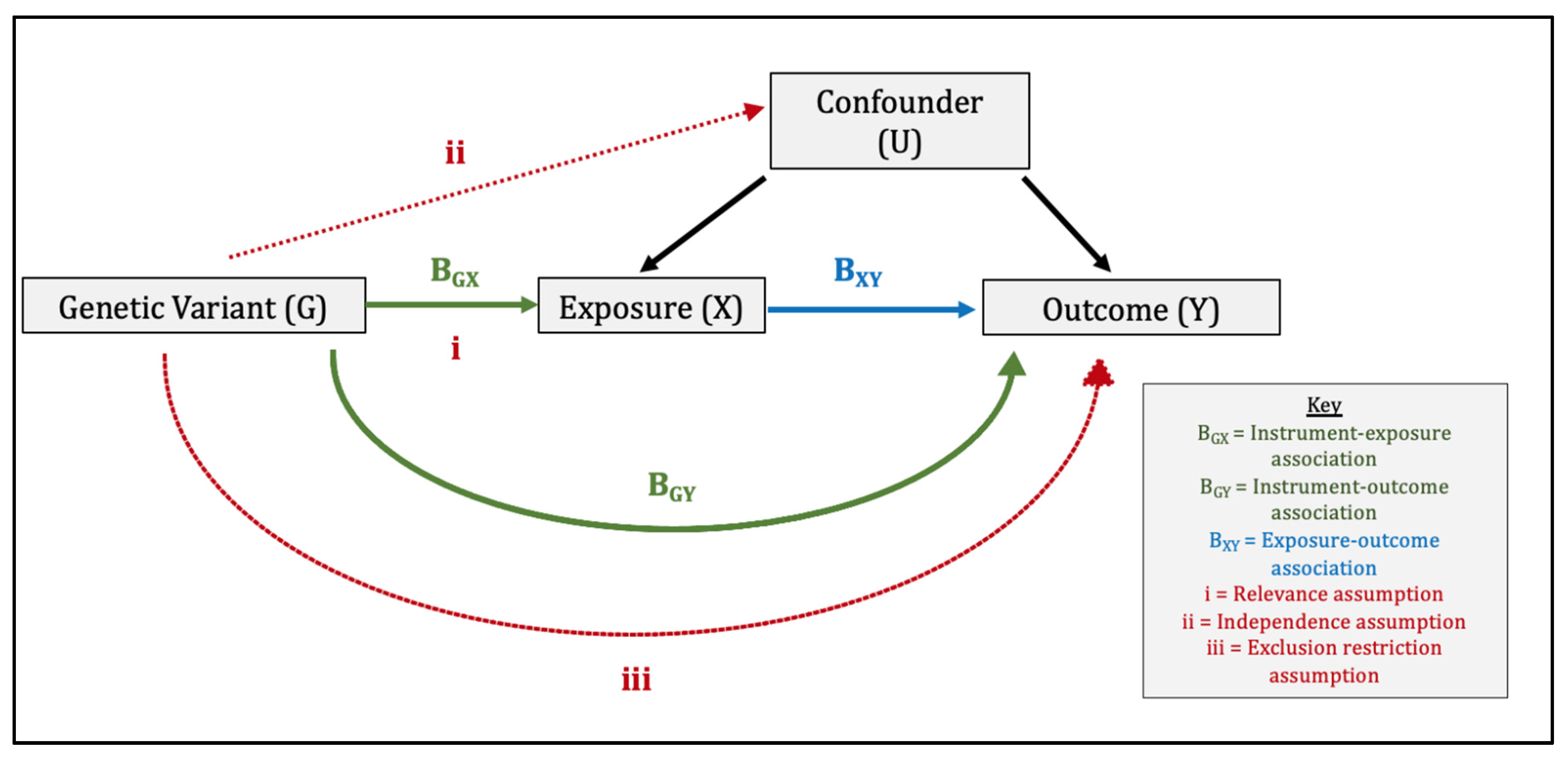{kind=link}
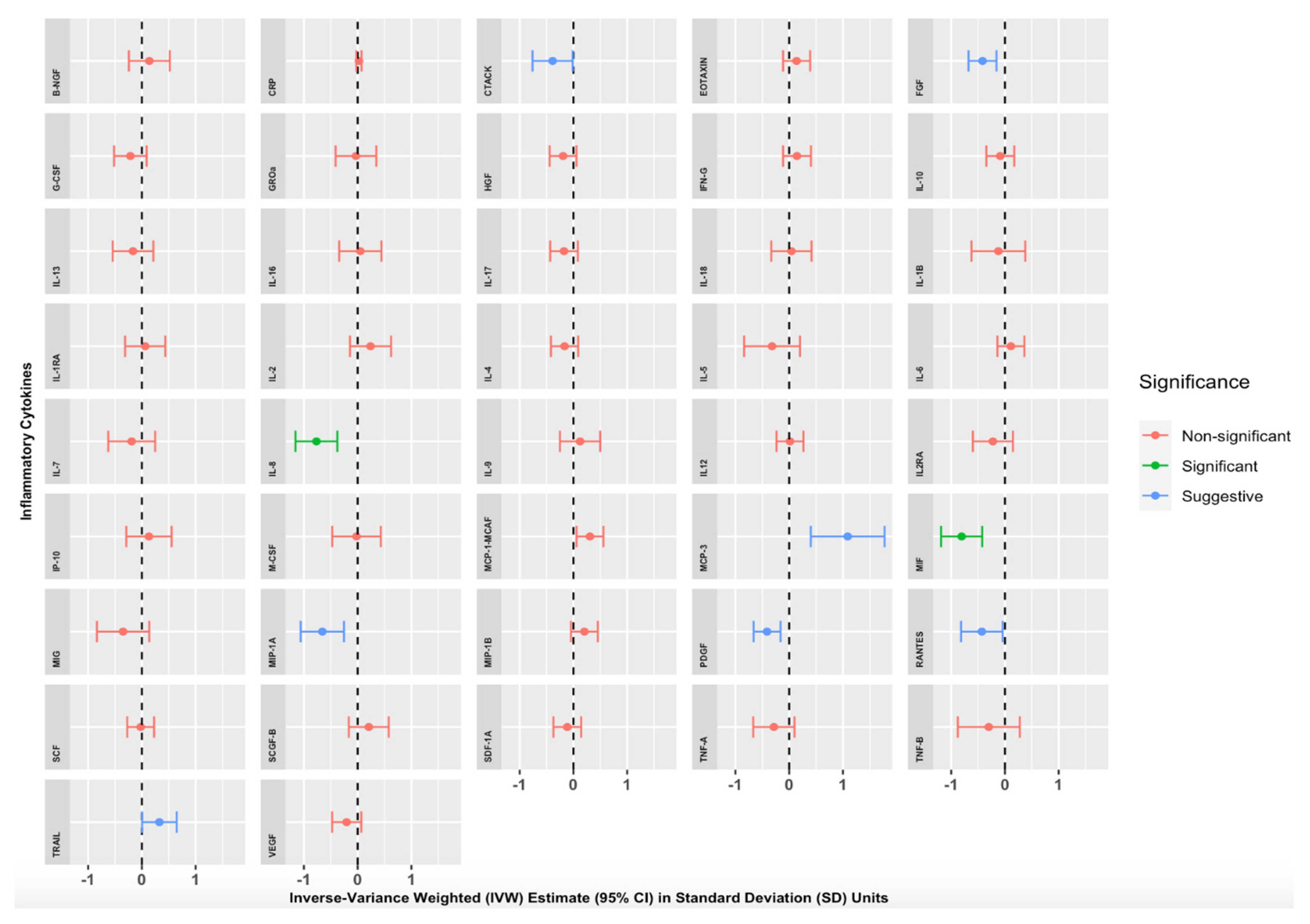{kind=link}
| Cytokine or Growth Factor |
|---|
| Beta-nerve growth factor (B-NGF) |
| Cutaneous T-cell-attracting chemokine (CTACK) |
| Eotaxin |
| Fibroblast growth factor 2 (FGF2) |
| Granulocyte-colony stimulating factor (G-CSF) |
| Growth regulated oncogene-alpha (GROa) |
| Hepatocyte growth factor (HGF) |
| Interferon gamma (IFN-G) |
| Interleukin 1 beta (IL-1B) |
| Interleukin 1 receptor alpha (IL-1RA) |
| Interleukin-2 (IL-2) |
| Interleukin-2 receptor alpha (IL-2RA) |
| Interleukin-4 (IL-4) |
| Interleukin-5 (IL-5) |
| Interleukin-6 (IL-6) |
| Interleukin-7 (IL-7) |
| Interleukin- 8 (IL-8) |
| Interleukin-9 (IL-9) |
| Interleukin-10 (IL-10) |
| Interleukin-12-P70 (IL-12-P70) |
| Interleukin-13 (IL-13) |
| Interleukin-16 (IL-16) |
| Interleukin-17 (IL-17) |
| Interleukin-18 (IL-18) |
| Interferon gamma-induced protein (IP-10) |
| Macrophage colony-stimulating factor(M-CSF) |
| Monocyte chemoattractant protein-1/Monocyte chemotactic and activating factor (MCP-1/MCAF) |
| Monocyte chemotactic protein-3 (MCP-3) |
| Macrophage migration inhibitory factor (MIF) |
| Mitogen-inducible-gene (MIG) |
| Macrophage inflammatory protein- 1 alpha (MIP-1A) |
| Macrophage inflammatory protein-1 beta (MIP-1B) |
| Platelet-derived growth factor (PDGF-BB) |
| Chemokine ligand 5 (RANTES) |
| Stem cell factor (SCF) |
| Stem cell growth factor- beta (SCGF-B) |
| Stromal cell-derived factor-1 alpha (SDF-1A) |
| Tumour necrosis factor-alpha (TNF-A) |
| Tumour necrosis factor-beta (TNF-B) |
| Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) |
| Vascular endothelial growth factor (VEGF) |
| C-reactive protein (CRP) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajasundaram, S.; Rahman, R.P.; Woolf, B.; Zhao, S.S.; Gill, D. Morning Cortisol and Circulating Inflammatory Cytokine Levels: A Mendelian Randomisation Study. Genes 2022, 13, 116. https://doi.org/10.3390/genes13010116
Rajasundaram S, Rahman RP, Woolf B, Zhao SS, Gill D. Morning Cortisol and Circulating Inflammatory Cytokine Levels: A Mendelian Randomisation Study. Genes. 2022; 13(1):116. https://doi.org/10.3390/genes13010116
Chicago/Turabian StyleRajasundaram, Skanda, Rezbieara P. Rahman, Benjamin Woolf, Sizheng Steven Zhao, and Dipender Gill. 2022. "Morning Cortisol and Circulating Inflammatory Cytokine Levels: A Mendelian Randomisation Study" Genes 13, no. 1: 116. https://doi.org/10.3390/genes13010116
APA StyleRajasundaram, S., Rahman, R. P., Woolf, B., Zhao, S. S., & Gill, D. (2022). Morning Cortisol and Circulating Inflammatory Cytokine Levels: A Mendelian Randomisation Study. Genes, 13(1), 116. https://doi.org/10.3390/genes13010116