
MEK Inhibition in a Newborn with RAF1-Associated Noonan Syndrome Ameliorates Hypertrophic Cardiomyopathy but Is Insufficient to Revert Pulmonary Vascular Disease

 ,
,  , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA Extraction
2.2. RNA Sequencing
2.3. Gene Expression Quantification
2.4. Gene Set Enrichment Analysis
3. Results
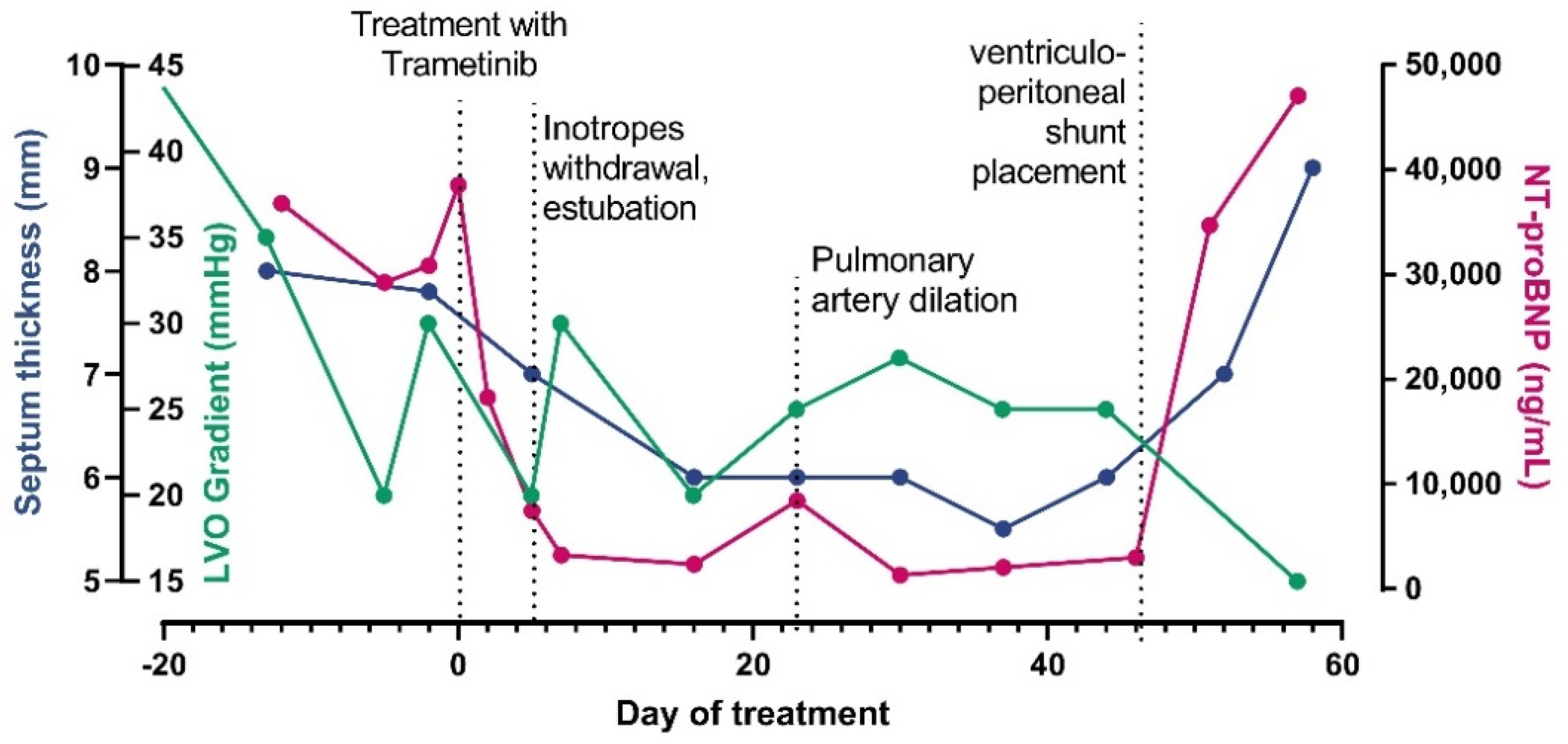
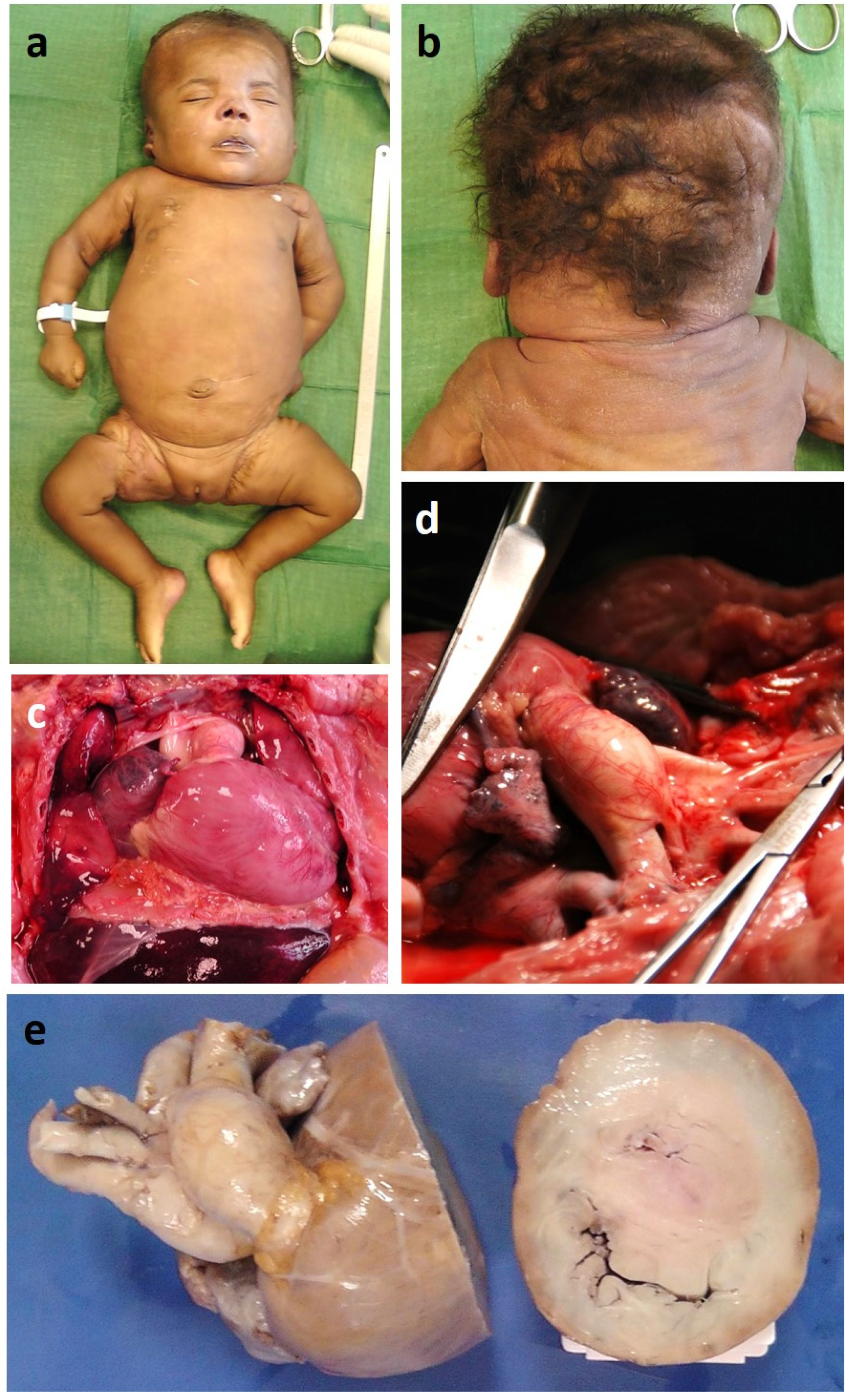
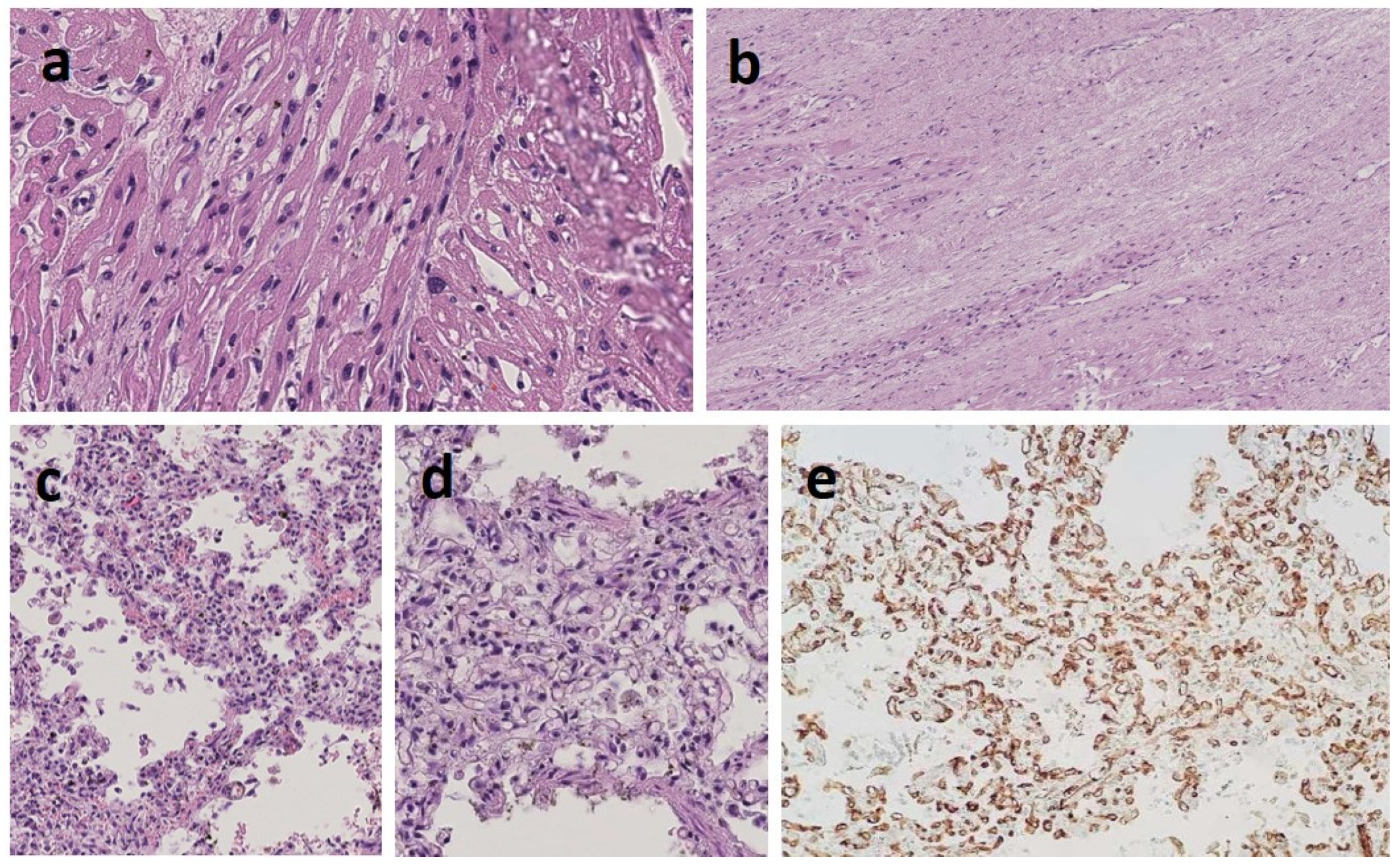
3.1. Clinical Report
3.2. Transcriptomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, M.; Zampino, G.; Gelb, B.D. Noonan syndrome: Clinical aspects and molecular pathogenesis. Mol. Syndromol. 2010, 1, 2–26. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, M.; Kalidas, K.; Shaw, A.; Song, X.; Musat, D.L.; van der Burgt, I.; Brunner, H.G.; Bertola, D.; Crosby, A.; Ion, A.; et al. PTPN11 mutations in Noonan syndrome: Molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 2002, 70, 1555–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, B.; Sarkozy, A.; Pennacchio, L.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007, 39, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Motta, M.; Giancotti, A.; Mastromoro, G.; Chandramouli, B.; Pinna, V.; Pantaleoni, F.; Di Giosaffatte, N.; Petrini, S.; Mazza, T.; D’Ambrosio, V.; et al. Clinical and functional characterization of a novel RASopathy-causing SHOC2 mutation associated with prenatal-onset hypertrophic cardiomyopathy. Hum. Mutat. 2019, 40, 1046–1056. [Google Scholar] [CrossRef]
- Aoki, Y.; Niihori, T.; Banjo, T.; Okamoto, N.; Mizuno, S.; Kurosawa, K.; Ogata, T.; Takada, F.; Yano, M.; Ando, T.; et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am. J. Hum. Genet. 2013, 93, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelb, B.D.; Roberts, A.E.; Tartaglia, M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog. Pediatr. Cardiol. 2015, 39, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Calcagni, G.; Adorisio, R.; Martinelli, S.; Grutter, G.; Baban, A.; Versacci, P.; Digilio, M.C.; Drago, F.; Gelb, B.D.; Tartaglia, M.; et al. Clinical Presentation and Natural History of Hypertrophic Cardiomyopathy in RASopathies. Heart Fail Clin. 2018, 14, 225–235. [Google Scholar] [CrossRef]
- Jaouadi, H.; Ben Chehida, A.; Kraoua, L.; Etchevers, H.C.; Argiro, L.; Kasdallah, N.; Blibech, S.; Delague, V.; Lévy, N.; Tebib, N.; et al. A severe clinical phenotype of Noonan syndrome with neonatal hypertrophic cardiomyopathy in the second case worldwide with RAF1 S259Y neomutation. Genet. Res. 2019, 101, e6. [Google Scholar] [CrossRef] [Green Version]
- Calcagni, G.; Limongelli, G.; D′Ambrosio, A.; Gesualdo, F.; Digilio, M.C.; Baban, A.; Albanese, S.; Versacci, P.; De Luca, E.; Ferrero, G.B.; et al. Cardiac defects, morbidity and mortality in patients affected by RASopathies. CARNET study results. Int. J. Cardiol. 2017, 245, 92–98. [Google Scholar] [CrossRef]
- Wu, X.; Simpson, J.; Hong, J.H.; Kim, K.-H.; Thavarajah, N.K.; Backx, P.H.; Neel, B.G.; Araki, T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Investig. 2011, 121, 1009–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andelfinger, G.; Marquis, C.; Raboisson, M.-J.; Théoret, Y.; Waldmüller, S.; Wiegand, G.; Gelb, B.D.; Zenker, M.; Delrue, M.-A.; Hofbeck, M. Hypertrophic Cardiomyopathy in Noonan Syndrome Treated by MEK-Inhibition. J. Am. Coll Cardiol. 2019, 73, 2237–2239. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Sun, X.; Kaufman, P.D. Ki-67: More than a proliferation marker. Chromosoma 2018, 127, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, G.B.; Picco, G.; Baldassarre, G.; Flex, E.; Isella, C.; Cantarella, D.; Corà, D.; Chiesa, N.; Crescenzio, N.; Timeus, F.; et al. Transcriptional hallmarks of noonan syndrome and noonan-like syndrome with loose anagen hair. Hum. Mutat. 2012, 33, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashley, E.A. Towards precision medicine. Nat Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Dhandapany, P.S.; Fabris, F.; Tonk, R.; Illaste, A.; Karakikes, I.; Sorourian, M.; Sheng, J.; Hajjar, R.J.; Tartaglia, M.; Sobie, E.A.; et al. Cyclosporine attenuates cardiomyocyte hypertrophy induced by RAF1 mutants in Noonan and LEOPARD syndromes. J. Mol. Cell. Cardiol. 2011, 51, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopper, R.K.; Feinstein, J.; Manning, M.A.; Benitz, W.; Hudgins, L. Neonatal pulmonary arterial hypertension and Noonan syndrome: Two fatal cases with a specificRAF1mutation. Am. J. Med. Genet. Part A 2015, 167, 882–885. [Google Scholar] [CrossRef]
- Thompson, D.; Patrick-Esteve, J.; Surcouf, J.W.; Rivera, D.; Castellanos, B.; Desai, P.; Lilje, C.; Lacassie, Y.; Marble, M.; Zambrano, R. RAF1 variants causing biventricular hypertrophic cardiomyopathy in two preterm infants: Further phenotypic delineation and review of literature. Clin. Dysmorphol. 2017, 26, 195–199. [Google Scholar] [CrossRef]
- Razzaque, A.; Nishizawa, T.; Komoike, Y.; Yagi, H.; Furutani, M.; Amo, R.; Kamisago, M.; Momma, K.; Katayama, H.; Nakagawa, M.; et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat. Genet. 2007, 39, 1013–1017. [Google Scholar] [CrossRef]
- Carcavilla, A.; Santomé, J.L.; Pinto, I.; Sánchez-Pozo, J.; Guillén-Navarro, E.; Martin-Frías, M.; Lapunzina, P.; Ezquieta, B. LEOPARD Syndrome: A Variant of Noonan Syndrome Strongly Associated with Hypertrophic Cardiomyopathy. Rev. Esp. Cardiol. 2013, 66, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Zarate, Y.A.; Lichty, A.W.; Champion, K.J.; Clarkson, L.K.; Holden, K.R.; Matheus, M.G. Unique Cerebrovascular Anomalies in Noonan Syndrome With RAF1 Mutation. J. Child Neurol. 2013, 29, NP13–NP17. [Google Scholar] [CrossRef]
- Gelb, B.D.; Tartaglia, M. Noonan Syndrome with Multiple Lentigines. 30 November 2007 [updated 14 May 2015]. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2021. [Google Scholar]
- Ko, J.M.; Kim, J.M.; Kim, G.H.; Yoo, H.W. PTPN11, SOS1, KRAS, and RAF1 gene analysis, and genotype-phenotype correlation in Korean patients with Noonan syndrome. J. Hum. Genet. 2008, 53, 999–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, J.; Lowe, A.M.; Salbert, B.A.; Sleeper, L.A.; Colan, S.D.; Cox, G.F.; Towbin, J.A.; Connuck, D.M.; Messere, J.E.; Lipshultz, S.E. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: A study from the Pediatric Cardiomyopathy Registry. Am. Heart J. 2012, 164, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Dhandapany, P.S.; Razzaque, A.; Muthusami, U.; Kunnoth, S.; Edwards, J.; Navarro, S.M.; Riess, I.; Pardo, S.; Sheng, J.; Rani, D.S.; et al. RAF1 mutations in childhood-onset dilated cardiomyopathy. Nat. Genet. 2014, 46, 635–639. [Google Scholar] [CrossRef]
- Brown, J.R.; Plotnick, G. Pulmonary Artery Aneurysm as a Cause for Chest Pain in a Patient with Noonan’s Syndrome: A Case Report. Cardiology 2007, 110, 249–251. [Google Scholar] [CrossRef]
- Purnell, R.; Williams, I.; Von Oppell, U.; Wood, A. Giant aneurysms of the sinuses of Valsalva and aortic regurgitation in a patient with Noonan’s syndrome. Eur. J. Cardiothorac. Surg. 2005, 28, 346–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, J.M.; Coupe, M.O.; Honey, M.; Miller, G.A. Aneurysms of the sinuses of Valsalva in Noonan′s syndrome. Eur. Heart J. 1989, 10, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Yanli, Z.; Xiaocong, W.; Liping, P.; Yan, M.; Wei, Y.; Shu, J. Diagnosis of a giant left atrial appendage aneurysm by contrast-enhanced echocardiography: Case report and literature review. J. Clin. Ultrasound 2020, 49, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, Y.; Fujimoto, N.; Ohashi, H.; Yamamoto, N.; Ito, H.; Mitani, Y.; Aoki, Y.; Imanaka-Yosida, K.; Ito, M.; Dohi, K. Case of Noonan Syndrome with an Expanding Coronary Arterial Aneurysm. Circ. Cardiovasc. Imaging 2019, 12, e009429. [Google Scholar] [CrossRef]
- Wong, C.K.; Cheng, C.H.; Lau, C.P.; Leung, W.H. Congenital coronary artery anomalies in Noonan’s syndrome. Am. Heart J. 1990, 119, 396–400. [Google Scholar] [CrossRef]
- Tahir, R.A.; Asmaro, K.; Pabaney, A.; Kole, M.; Nypaver, T.; Marin, H. Republished: Separate origins of the left internal and external carotid arteries from the aortic arch and cervical internal carotid artery aneurysm in a patient with Noonan syndrome. J. NeuroInterventional Surg. 2016, 9, e11. [Google Scholar] [CrossRef]
- Weatherald, J.; Dorfmüller, P.; Perros, F.; Ghigna, M.-R.; Girerd, B.; Humbert, M.; Montani, D. Pulmonary capillary haemangiomatosis: A distinct entity? Eur. Respir. Rev. 2020, 29, 190168. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, W.B.; Choi, J.H.; Lee, S.N. A Case of Congenital Pulmonary Lymphangiectasia in Noonan Syndrome. J Korean Pediatr. Soc. 1997, 40, 877–882. [Google Scholar]
- Puvabanditsin, S.; Abellar, R.; Madubuko, A.; Mehta, R.; Walzer, L. Pulmonary Vasculitis and a Horseshoe Kidney in Noonan Syndrome. Case Rep. Pathol. 2018, 2018, 6829586. [Google Scholar] [CrossRef] [Green Version]
- Guignabert, C.; Tu, L.; Le Hiress, M.; Ricard, N.; Sattler, C.; Seferian, A.; Huertas, A.; Humbert, M.; Montani, D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur. Respir. Rev. 2013, 22, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, K.S.; Elinoff, J.M.; Wang, S.; Gairhe, S.; Ferreyra, G.A.; Cai, R.; Sun, J.; Solomon, M.A.; Danner, R.L. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L187–L201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oviedo, A.; Abramson, L.P.; Worthington, R.; Dainauskas, J.R.; Crawford, S.E. Congenital pulmonary capillary hemangiomatosis: Report of two cases and review of the literature. Pediatr. Pulmonol. 2003, 36, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Luo, J.; Hou, N. Molecular Mechanism of Hippo-YAP1/TAZ Pathway in Heart Development, Disease, and Regeneration. Front. Physiol. 2020, 11, 389. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-S.; Park, J.-H.; Lim, H.-J.; Park, H.-Y. HB-EGF induces cardiomyocyte hypertrophy via an ERK5-MEF2A-COX2 signaling pathway. Cell. Signal. 2011, 23, 1100–1109. [Google Scholar] [CrossRef]
- López, B.; González, A.; Lindner, D.; Westermann, D.; Ravassa, S.; Beaumont, J.; Gallego, I.; Zudaire, A.; Brugnolaro, C.; Querejeta, R.; et al. Osteopontin-mediated myocardial fibrosis in heart failure: A role for lysyl oxidase? Cardiovasc. Res. 2013, 99, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lim, S.T.; Teo, M.H.Y.; Tan, M.S.Y.; Kulkarni, M.D.; Qiu, B.; Li, A.; Lal, S.; Remedios, C.G.; Tan, N.S.; et al. Collaborative Regulation of LRG1 by TGF-β1 and PPAR-β/δ Modulates Chronic Pressure Overload-Induced Cardiac Fibrosis. Circ. Heart Fail. 2019, 12, e005962. [Google Scholar] [CrossRef]
- Gratzinger, D.; Zhao, S.; West, R.; Rouse, R.V.; Vogel, H.; Gil, E.C.; Levy, R.; Lossos, I.S.; Natkunam, Y. The Transcription Factor LMO2 Is a Robust Marker of Vascular Endothelium and Vascular Neoplasms and Selected Other Entities. Am. J. Clin. Pathol. 2009, 131, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Leong, Z.P.; Okida, A.; Higuchi, M.; Yamano, Y.; Hikasa, Y. Reversal effects of low-dose imatinib compared with sunitinib on monocrotaline-induced pulmonary and right ventricular remodeling in rats. Vasc. Pharmacol. 2018, 100, 41–50. [Google Scholar] [CrossRef]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mussa, A.; Carli, D.; Giorgio, E.; Villar, A.M.; Cardaropoli, S.; Carbonara, C.; Campagnoli, M.F.; Galletto, P.; Palumbo, M.; Olivieri, S.; et al. MEK Inhibition in a Newborn with RAF1-Associated Noonan Syndrome Ameliorates Hypertrophic Cardiomyopathy but Is Insufficient to Revert Pulmonary Vascular Disease. Genes 2022, 13, 6. https://doi.org/10.3390/genes13010006
Mussa A, Carli D, Giorgio E, Villar AM, Cardaropoli S, Carbonara C, Campagnoli MF, Galletto P, Palumbo M, Olivieri S, et al. MEK Inhibition in a Newborn with RAF1-Associated Noonan Syndrome Ameliorates Hypertrophic Cardiomyopathy but Is Insufficient to Revert Pulmonary Vascular Disease. Genes. 2022; 13(1):6. https://doi.org/10.3390/genes13010006
Chicago/Turabian StyleMussa, Alessandro, Diana Carli, Elisa Giorgio, Anna Maria Villar, Simona Cardaropoli, Caterina Carbonara, Maria Francesca Campagnoli, Paolo Galletto, Martina Palumbo, Simone Olivieri, and et al. 2022. "MEK Inhibition in a Newborn with RAF1-Associated Noonan Syndrome Ameliorates Hypertrophic Cardiomyopathy but Is Insufficient to Revert Pulmonary Vascular Disease" Genes 13, no. 1: 6. https://doi.org/10.3390/genes13010006
APA StyleMussa, A., Carli, D., Giorgio, E., Villar, A. M., Cardaropoli, S., Carbonara, C., Campagnoli, M. F., Galletto, P., Palumbo, M., Olivieri, S., Isella, C., Andelfinger, G., Tartaglia, M., Botta, G., Brusco, A., Medico, E., & Ferrero, G. B. (2022). MEK Inhibition in a Newborn with RAF1-Associated Noonan Syndrome Ameliorates Hypertrophic Cardiomyopathy but Is Insufficient to Revert Pulmonary Vascular Disease. Genes, 13(1), 6. https://doi.org/10.3390/genes13010006