
Capturing SNP Association across the NK Receptor and HLA Gene Regions in Multiple Sclerosis by Targeted Penalised Regression Models

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
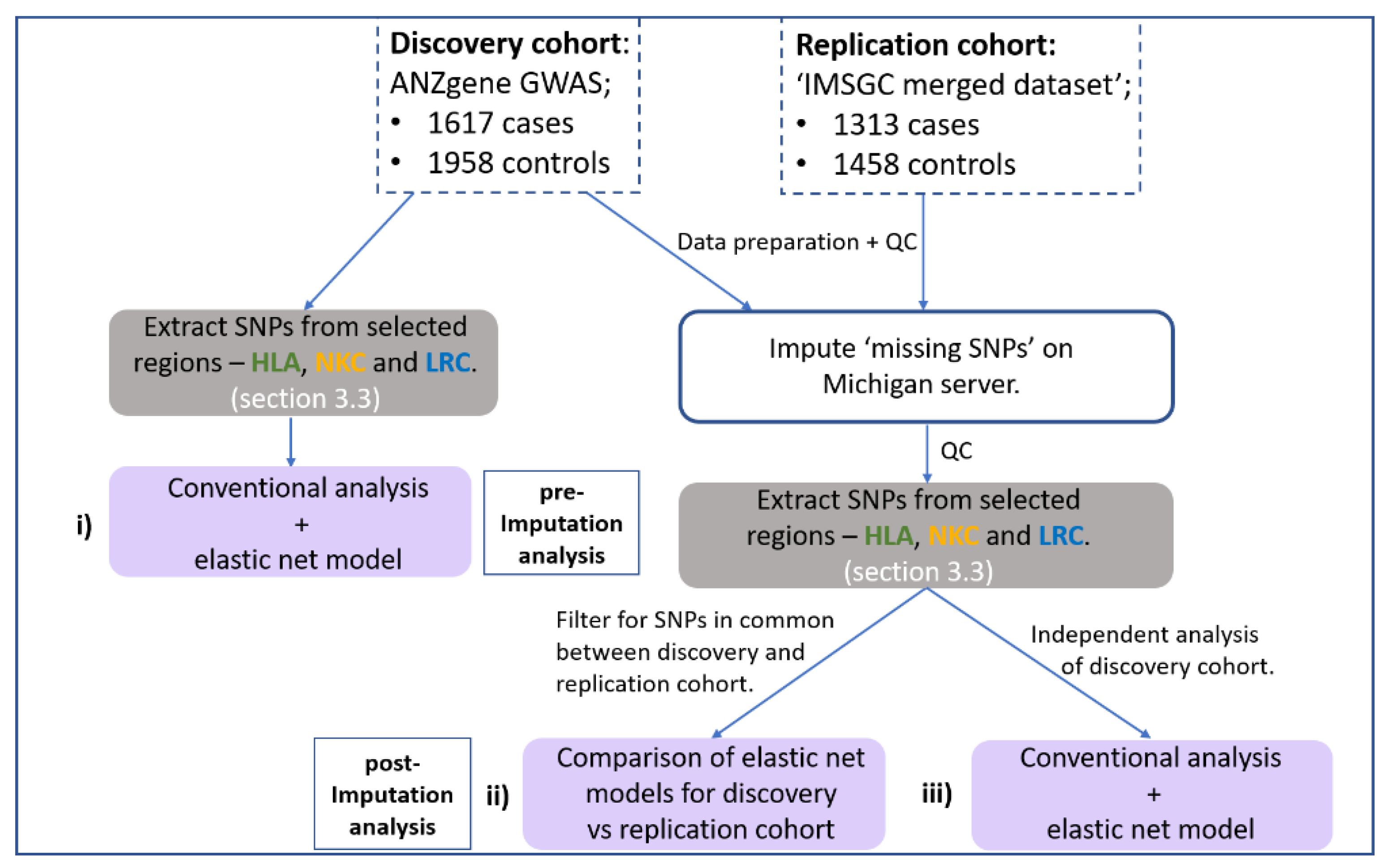
2.1. Overview of Analysis Pipeline
2.2. Datasets and QC
2.3. Imputation
2.4. SNP Boundary Selection and Extraction
2.5. Standard Association Testing
2.6. ‘Elastic Net Model’ Optimisation with Stability Selection via BootNet (Iterative Subsampling)
2.7. Replication Analysis Using SNPs in Common
2.8. Independent Analysis of the Discovery Cohort
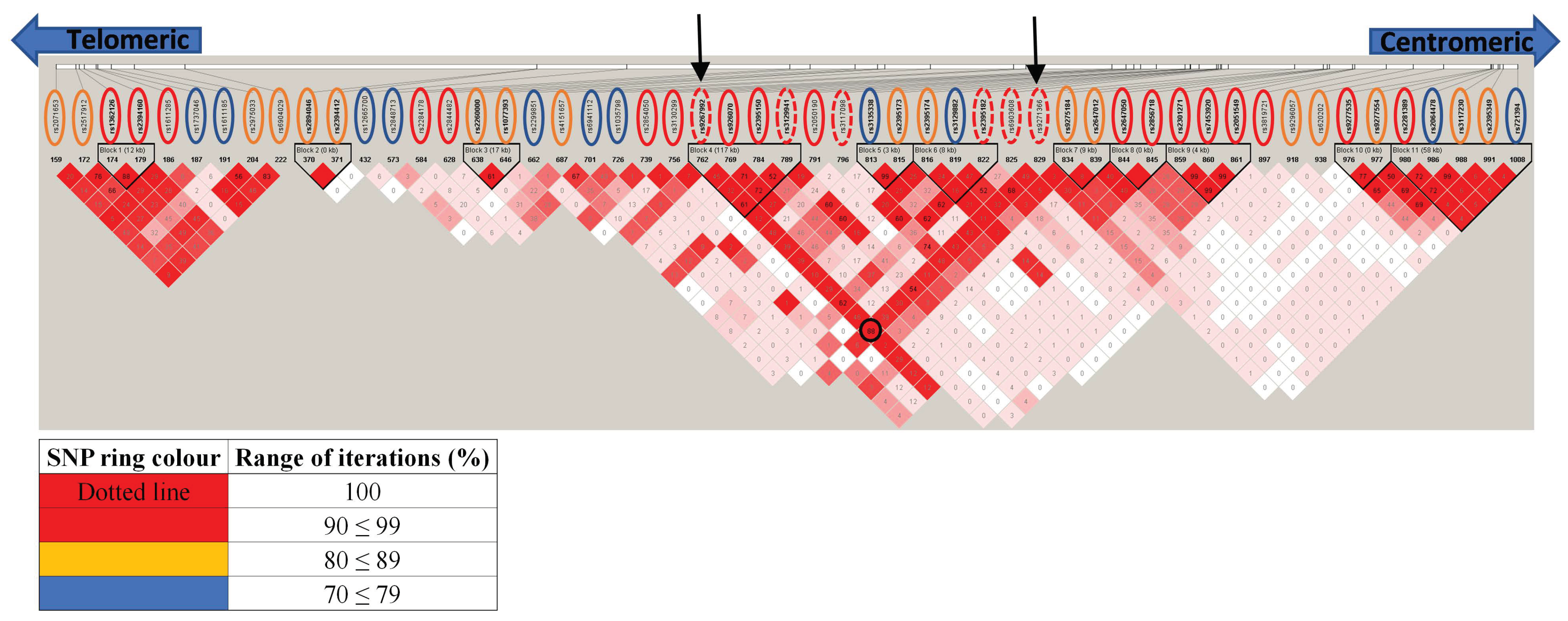
2.9. Haploview Analysis
2.10. Epistasis Analysis
2.11. Code Availability
3. Results
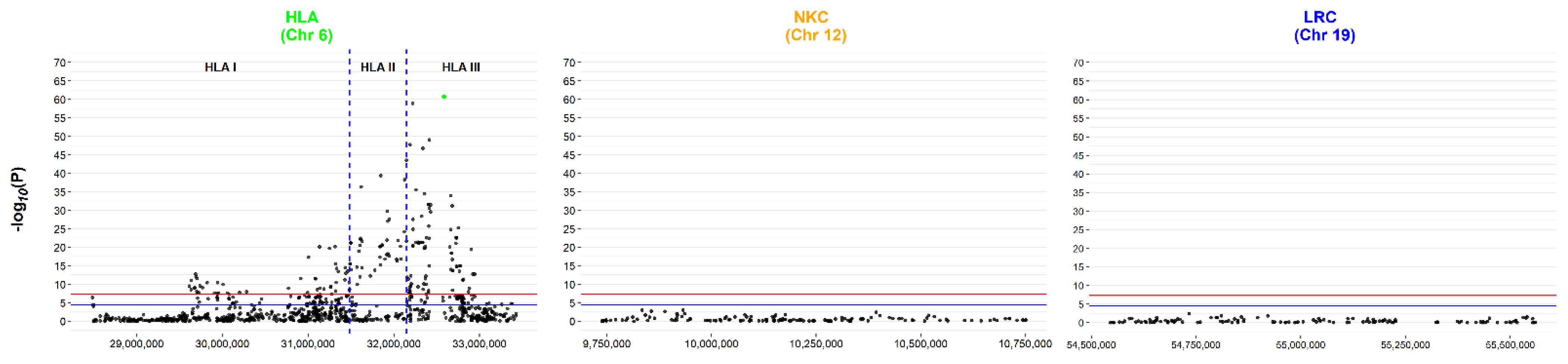
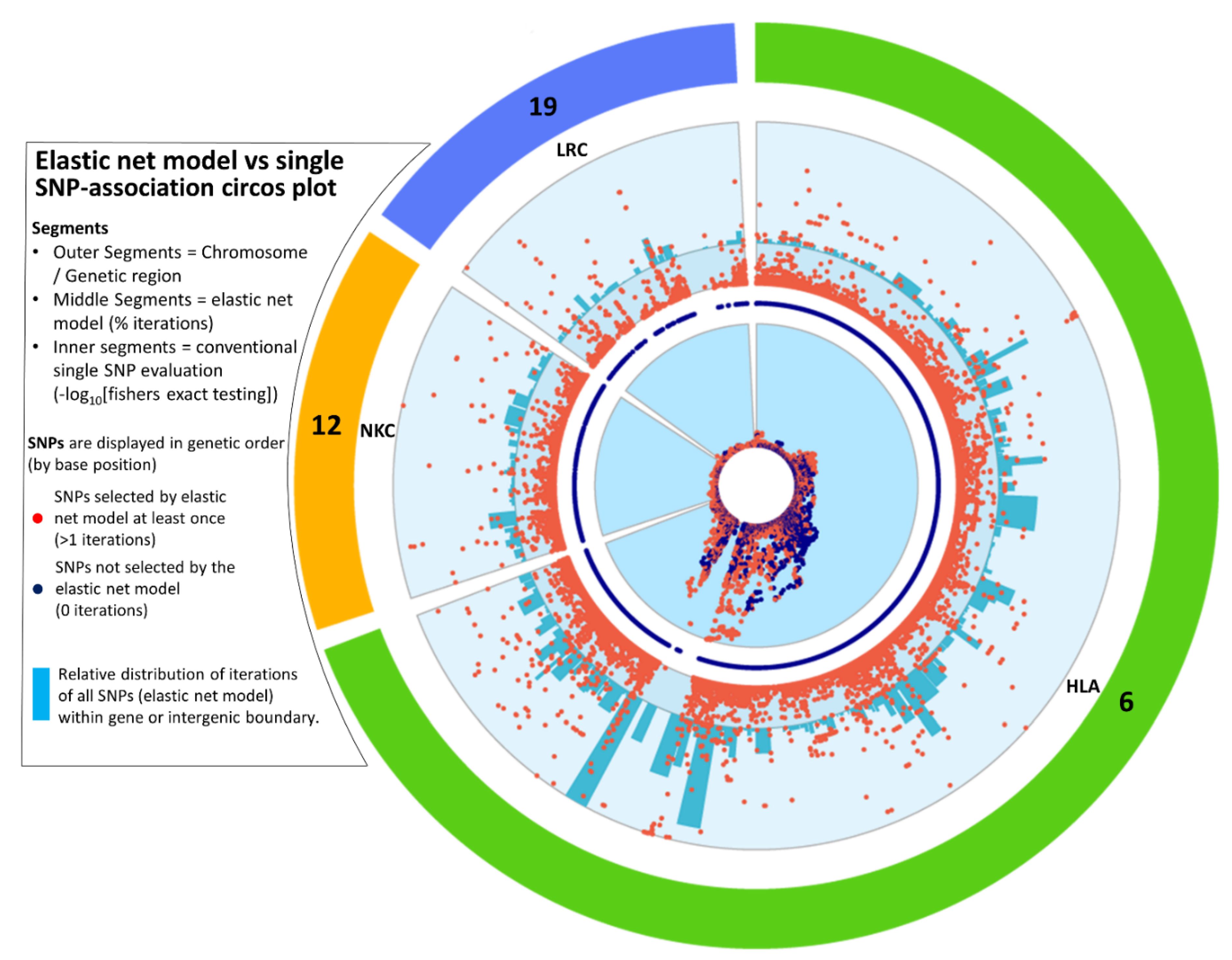
3.1. Re-analysis of the ANZgene GWAS by Elastic Net Identifies SNPs above and below Conventional p-Value Thresholds
3.2. Replication Analysis Using SNPs Common to Both the Discovery and Replication Cohort
3.3. Independent Analysis of Discovery Cohort Provides Further Insight Due to Imputation
3.4. Epistasis Analysis
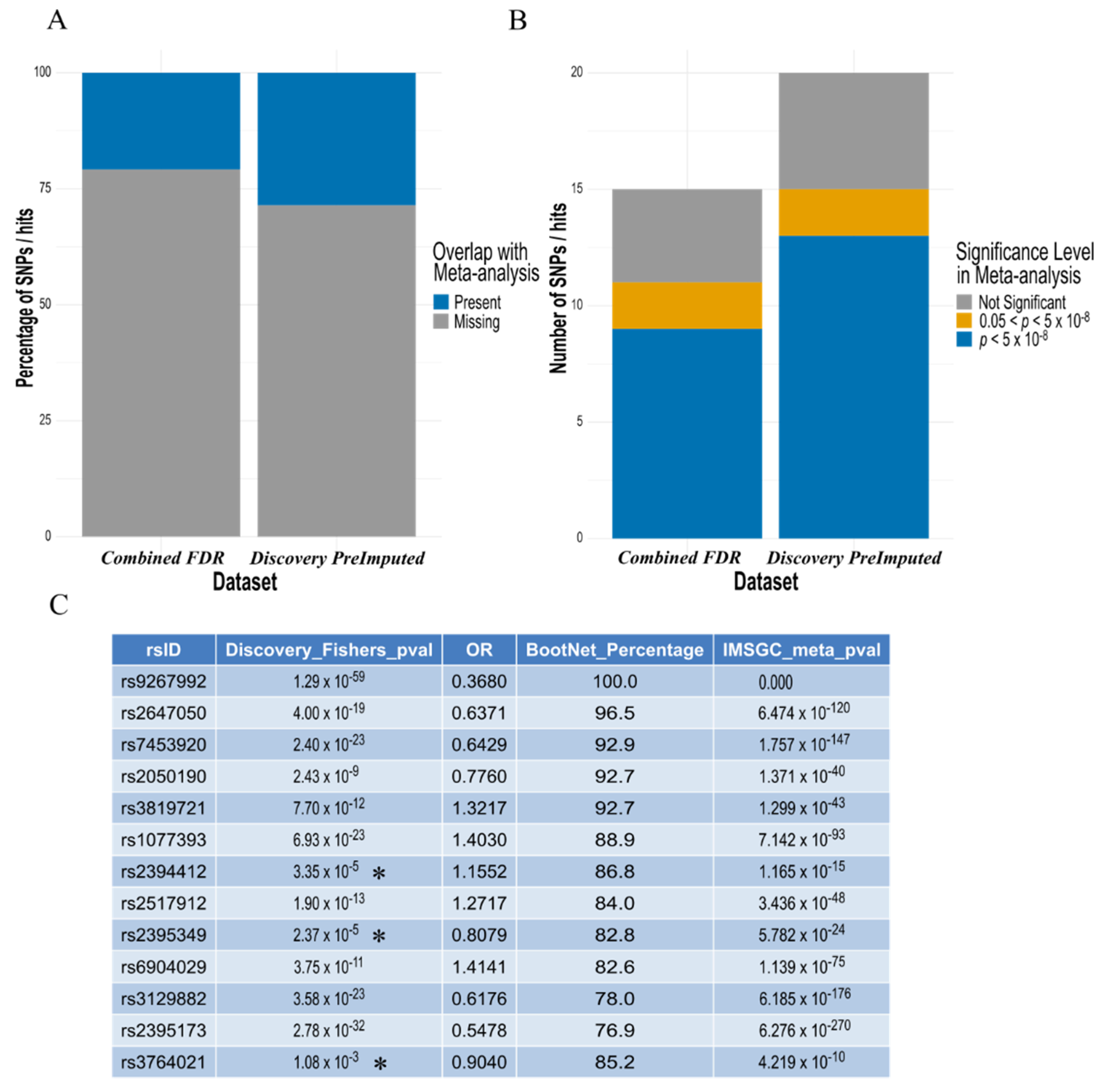
3.5. Confirmation of Elastic Net Model Hits to Largest MS Meta-Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tarlinton, R.E.; Khaibullin, T.; Granatov, E.; Martynova, E.; Rizvanov, A.; Khaiboullina, S. The Interaction between Viral and Environmental Risk Factors in the Pathogenesis of Multiple Sclerosis. Int. J. Mol. Sci. 2019, 20, 303. [Google Scholar] [CrossRef] [Green Version]
- Simpson, S., Jr.; van der Mei, I.; Lucas, R.M.; Ponsonby, A.L.; Broadley, S.; Blizzard, L.; Taylor, B. Sun Exposure across the Life Course Significantly Modulates Early Multiple Sclerosis Clinical Course. Front. Neurol. 2018, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wang, R.; Li, Z.; Wang, Y.; Gao, C.; Lv, X.; Song, Y.; Li, B. The risk of smoking on multiple sclerosis: A meta-analysis based on 20,626 cases from case-control and cohort studies. PeerJ 2016, 4, e1797. [Google Scholar] [CrossRef] [PubMed]
- Burnard, S.; Lechner-Scott, J.; Scott, R.J. EBV and MS: Major cause, minor contribution or red-herring? Mult. Scler. Relat. Disord. 2017, 16, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzaraki, V.; Kumar, V.; Wijmenga, C.; Zhernakova, A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, J.A.; Oksenberg, J.R. The immunogenetics of multiple sclerosis: A comprehensive review. J. Autoimmun. 2015, 64, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Roche, P.A.; Cresswell, P. Antigen Processing and Presentation Mechanisms in Myeloid Cells. Microbiol. Spectr. 2016, 4, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Traherne, J.A. Human MHC architecture and evolution: Implications for disease association studies. Int. J. Immunogenet. 2008, 35, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Howell, W.M. HLA and disease: Guilt by association. Int. J. Immunogenet. 2014, 41, 1–12. [Google Scholar] [CrossRef]
- Yokoyama, W.M.; Plougastel, B.F.M. Immune functions encoded by the natural killer gene complex. Nat. Rev. Immunol. 2003, 3, 304–316. [Google Scholar] [CrossRef]
- Barrow, A.D.; Trowsdale, J. The extended human leukocyte receptor complex: Diverse ways of modulating immune responses. Immunol. Rev. 2008, 224, 98–123. [Google Scholar] [CrossRef]
- Kelley, J.; Walter, L.; Trowsdale, J. Comparative Genomics of Natural Killer Cell Receptor Gene Clusters. PLoS Genet. 2005, 1, e27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruana, P.; Lemmert, K.; Ribbons, K.; Lea, R.; Lechner-Scott, J. Natural killer cell subpopulations are associated with MRI activity in a relapsing-remitting multiple sclerosis patient cohort from Australia. Mult. Scler. 2016, 23, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Chanvillard, C.; Jacolik, R.F.; Infante-Duarte, C.; Nayak, R.C. The role of natural killer cells in multiple sclerosis and their therapeutic implications. Front. Immunol. 2013, 4, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Rodriguez, J.E.; Lopez-Botet, M.; Munteis, E.; Rio, J.; Roquer, J.; Montalban, X.; Comabella, M. Natural killer cell phenotype and clinical response to interferon-beta therapy in multiple sclerosis. Clin. Immunol. 2011, 141, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Darlington, P.J.; Stopnicki, B.; Touil, T.; Doucet, J.-S.; Fawaz, L.; Roberts, M.E.; Boivin, M.-N.; Arbour, N.; Freedman, M.S.; Atkins, H.L.; et al. Natural Killer Cells Regulate Th17 Cells After Autologous Hematopoietic Stem Cell Transplantation for Relapsing Remitting Multiple Sclerosis. Front. Immunol. 2018, 9, 834. [Google Scholar] [CrossRef]
- Poggi, A.; Zocchi, M.R. NK cell autoreactivity and autoimmune diseases. Front. Immunol. 2014, 5, 27. [Google Scholar] [CrossRef]
- Schleinitz, N.; Vély, F.; Harlé, J.-R.; Vivier, E. Natural killer cells in human autoimmune diseases. Immunology 2010, 131, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Morse, R.H.; Seguin, R.; McCrea, E.L.; Antel, J.P. NK cell-mediated lysis of autologous human oligodendrocytes. J. Neuroimmunol. 2001, 116, 107–115. [Google Scholar] [CrossRef]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Rünzi, A.; Kuhlmann, T.; Posevitz-Fejfár, A.; Schwab, N.; Schneider-Hohendorf, T.; Herich, S.; Held, K.; Konjević, M.; et al. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2973. [Google Scholar] [CrossRef] [Green Version]
- Parnell, G.P.; Booth, D.R. The Multiple Sclerosis (MS) Genetic Risk Factors Indicate both Acquired and Innate Immune Cell Subsets Contribute to MS Pathogenesis and Identify Novel Therapeutic Opportunities. Front. Immunol. 2017, 8, 425. [Google Scholar] [CrossRef]
- Canto, E.; Oksenberg, J.R. Multiple sclerosis genetics. Mult. Scler. J. 2018, 24, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.; Donnelly, P.; The International HapMap, C. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar] [CrossRef] [Green Version]
- Baranzini, S.E.; Galwey, N.W.; Wang, J.; Khankhanian, P.; Lindberg, R.; Pelletier, D.; Wu, W.; Uitdehaag, B.M.J.; Kappos, L.; Gene, M.S.A.C.; et al. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Genet. 2009, 18, 2078–2090. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 22. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.-H.; Wang, Q.; Bolla, M.K.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.T.; Chen, Y.F.; Hastie, T.; Sobel, E.; Lange, K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics 2009, 25, 714–721. [Google Scholar] [CrossRef]
- Waldmann, P.; Ferenčaković, M.; Mészáros, G.; Khayatzadeh, N.; Curik, I.; Sölkner, J. AUTALASSO: An automatic adaptive LASSO for genome-wide prediction. BMC Bioinform. 2019, 20, 167. [Google Scholar] [CrossRef]
- Qian, J.; Du, W.; Tanigawa, Y.; Aguirre, M.; Tibshirani, R.; Rivas, M.A.; Hastie, T. A Fast and Flexible Algorithm for Solving the Lasso in Large-scale and Ultrahigh-dimensional Problems. bioRxiv 2019, 630079. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, P.; Mészáros, G.; Gredler, B.; Fürst, C.; Sölkner, J. Evaluation of the lasso and the elastic net in genome-wide association studies. Front. Genet. 2013, 4, 270. [Google Scholar] [CrossRef] [Green Version]
- Benton, M.C.; Sutherland, H.G.; Macartney-Coxson, D.; Haupt, L.M.; Lea, R.A.; Griffiths, L.R. Methylome-wide association study of whole blood DNA in the Norfolk Island isolate identifies robust loci associated with age. Aging 2017, 9, 753–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meinshausen, N.; Bühlmann, P. Stability selection. J. R. Stat. Soc. 2010, 72, 417–473. [Google Scholar] [CrossRef]
- CONSORTIUM, I.M.S.G. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef] [Green Version]
- Fry, B.; Maller, J.; Barrett, J.C.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2004, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- The Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene). Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet. 2009, 41, 824–828. [Google Scholar] [CrossRef]
- De Jager, P.L.; Jia, X.; Wang, J.; de Bakker, P.I.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009, 41, 776–782. [Google Scholar] [CrossRef] [Green Version]
- Hafler, D.A.; Compston, A.; Sawcer, S.; Lander, E.S.; Daly, M.J.; De Jager, P.L.; de Bakker, P.I.; Gabriel, S.B.; Mirel, D.B.; Ivinson, A.J.; et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007, 357, 851–862. [Google Scholar] [CrossRef] [Green Version]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- A Language and Environment for Statistical Computing; Version R 3.5.1; R Foundation for Statistical Computing: Vienna, Austria, 2018.
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- PLINK. Version 1.9. Christopher Chang: Berkeley, CA, USA. 2018. Available online: www.cog-genomics.org/plink/1.9/ (accessed on 27 June 2020).
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, P.-R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, Y.A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, A.I.; van der Lee, S.J.; Bonnemaijer, P.W.M.; Hohn, R.; Nag, A.; Gharahkhani, P.; Khawaja, A.P.; Broer, L.; Foster, P.J.; Hammond, C.J.; et al. Haplotype reference consortium panel: Practical implications of imputations with large reference panels. Hum. Mutat. 2017, 38, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Genome Reference Consortium. Available online: https://www.ncbi.nlm.nih.gov/grc (accessed on 1 June 2019).
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv 2014. [Google Scholar] [CrossRef]
- Glusman, G.; Caballero, J.; Mauldin, D.E.; Hood, L.; Roach, J.C. Kaviar: An accessible system for testing SNV novelty. Bioinformatics 2011, 27, 3216–3217. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef] [PubMed] [Green Version]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, X.; Luo, H.; Fan, Z.; Luo, J.; He, S.; Yue, H.; Zhang, P.; Chen, R. BioCircos.js: An interactive Circos JavaScript library for biological data visualization on web applications. Bioinformatics 2016, 32, 1740–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BioCircos: Interactive Circular Visualization of Genomic Data Using ’htmlwidgets’ and ’BioCircos.js; Version 0.3.4; Erich Neuwirth: Vienna, Austria, 2019.
- Benton, M.; Blick, R.; White, N.; Burnard, S. Sirselim/bootnet: First Release to Coincide with MS Manuscript Publication. 2020. Available online: https://doi.org/10.5281/zenodo.3966550 (accessed on 10 November 2021).
- Plant, S.R.; Arnett, H.A.; Ting, J.P.-Y. Astroglial-derived lymphotoxin-α exacerbates inflammation and demyelination, but not remyelination. Glia 2005, 49, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Patsopoulos, N.A.; Barcellos, L.F.; Hintzen, R.Q.; Schaefer, C.; van Duijn, C.M.; Noble, J.A.; Raj, T.; Gourraud, P.-A.; IMSGC; ANZgene; et al. Fine-Mapping the Genetic Association of the Major Histocompatibility Complex in Multiple Sclerosis: HLA and Non-HLA Effects. PLOS Genet. 2013, 9, e1003926. [Google Scholar] [CrossRef]
- Maltby, V.E.; Lea, R.A.; Graves, M.C.; Sanders, K.A.; Benton, M.C.; Tajouri, L.; Scott, R.J.; Lechner-Scott, J. Genome-wide DNA methylation changes in CD19+ B cells from relapsing-remitting multiple sclerosis patients. Sci. Rep. 2018, 8, 17418. [Google Scholar] [CrossRef]
- Christensen, J.R.; Börnsen, L.; Hesse, D.; Krakauer, M.; Sørensen, P.S.; Søndergaard, H.B.; Sellebjerg, F. Cellular sources of dysregulated cytokines in relapsing-remitting multiple sclerosis. J. Neuroinflamm. 2012, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Wang, K.; Qu, H.-Q.; Zhang, H.; Bradfield, J.; Kim, C.; Frackleton, E.; Hou, C.; Glessner, J.T.; Chiavacci, R.; et al. From Disease Association to Risk Assessment: An Optimistic View from Genome-Wide Association Studies on Type 1 Diabetes. PLoS Genet. 2009, 5, e1000678. [Google Scholar] [CrossRef]
- Park, M.Y.; Hastie, T. Penalized logistic regression for detecting gene interactions. Biostatistics 2008, 9, 30–50. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Taheri, M.; Omrani, M.D.; Daaee, A.; Mohammad-Rahimi, H. Application of Artificial Neural Network for Prediction of Risk of Multiple Sclerosis Based on Single Nucleotide Polymorphism Genotypes. J. Mol. Neurosci. 2020, 70, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Arloth, J.; Eraslan, G.; Andlauer, T.F.M.; Martins, J.; Iurato, S.; Kühnel, B.; Waldenberger, M.; Frank, J.; Gold, R.; Hemmer, B.; et al. DeepWAS: Multivariate genotype-phenotype associations by directly integrating regulatory information using deep learning. PLOS Comput. Biol. 2020, 16, e1007616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltby, V.E.; Graves, M.C.; Lea, R.A.; Benton, M.C.; Sanders, K.A.; Tajouri, L.; Scott, R.J.; Lechner-Scott, J. Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin. Epigenet. 2015, 7, 118. [Google Scholar] [CrossRef] [Green Version]
- Graves, M.C.; Benton, M.; Lea, R.A.; Boyle, M.; Tajouri, L.; Macartney-Co×son, D.; Scott, R.J.; Lechner-Scott, J. Methylation differences at the HLA-DRB1 locus in CD4+ T-Cells are associated with multiple sclerosis. Mult. Scler. 2014, 20, 1033–1041. [Google Scholar] [CrossRef]
- Bánlaki, Z.; Szabó, J.A.; Szilágyi, Á.; Patócs, A.; Prohászka, Z.; Füst, G.; Doleschall, M. Intraspecific evolution of human RCC× copy number variation traced by haplotypes of the CYP21A2 gene. Genome Biol. Evol. 2013, 5, 98–112. [Google Scholar] [CrossRef] [Green Version]
- Tassabehji, M.; Strachan, T.; Anderson, M.; Campbell, R.D.; Collier, S.; Lako, M. Identification of a novel family of human endogenous retroviruses and characterization of one family member, HERV-K(C4), located in the complement C4 gene cluster. Nucleic Acids Res. 1994, 22, 5211–5217. [Google Scholar] [CrossRef] [Green Version]
- Caillier, S.J.; Briggs, F.; Cree, B.A.C.; Baranzini, S.E.; Fernandez-Viña, M.; Ramsay, P.P.; Khan, O.; Royal, W., III; Hauser, S.L.; Barcellos, L.F.; et al. Uncoupling the roles of HLA-DRB1 and HLA-DRB5 genes in multiple sclerosis. J. Immunol. 2008, 181, 5473–5480. [Google Scholar] [CrossRef] [Green Version]
- Tatomir, A.; Talpos-Caia, A.; Anselmo, F.; Kruszewski, A.M.; Boodhoo, D.; Rus, V.; Rus, H. The complement system as a biomarker of disease activity and response to treatment in multiple sclerosis. Immunol. Res. 2017, 65, 1103–1109. [Google Scholar] [CrossRef]
- Ingram, G.; Hakobyan, S.; Robertson, N.P.; Morgan, B.P. Complement in multiple sclerosis: Its role in disease and potential as a biomarker. Clin. Exp. Immunol. 2009, 155, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Groen, K.; Maltby, V.E.; Sanders, K.A.; Scott, R.J.; Tajouri, L.; Lechner-Scott, J. Erythrocytes in multiple sclerosis—forgotten contributors to the pathophysiology? Mult. Scler. J.-Exp. Transl. Clin. 2016, 2, 2055217316649981. [Google Scholar] [CrossRef] [PubMed]
- Mellqvist, U.H.; Hansson, M.; Brune, M.; Dahlgren, C.; Hermodsson, S.; Hellstrand, K. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: Role of reactive oxygen species and regulation by histamine. Blood 2000, 96, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Matsunaga, K. Susceptibility of natural killer (NK) cells to reactive o×ygen species (ROS) and their restoration by the mimics of superoxide dismutase (SOD). Cancer Biother. Radiopharm. 1998, 13, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Malorni, W.; Straface, E.; Genova, G.D.; Fattorossi, A.; Rivabene, R.; Camponeschi, B.; Masella, R.; Viora, M. Oxidized Low-Density Lipoproteins Affect Natural Killer Cell Activity by Impairing Cytoskeleton Function and Altering the Cytokine Network. Exp. Cell Res. 1997, 236, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Kastrukoff, L.F.; Morgan, N.G.; Zecchini, D.; White, R.; Petkau, A.J.; Satoh, J.; Paty, D.W. A role for natural killer cells in the immunopathogenesis of multiple sclerosis. J. Neuroimmunol. 1998, 86, 123–133. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Lau, A.; Wee, R.; Zecchini, D.; White, R.; Paty, D.W. Clinical relapses of multiple sclerosis are associated with ‘novel’ valleys in natural killer cell functional activity. J. Neuroimmunol. 2003, 145, 103–114. [Google Scholar] [CrossRef]
- Gironi, M.; Borgiani, B.; Mariani, E.; Cursano, C.; Mendozzi, L.; Cavarretta, R.; Saresella, M.; Clerici, M.; Comi, G.; Rovaris, M.; et al. Oxidative stress is differentially present in multiple sclerosis courses, early evident, and unrelated to treatment. J. Immunol. Res. 2014, 2014, 961863. [Google Scholar] [CrossRef] [Green Version]
- Gustavsen, M.W.; Viken, M.K.; Celius, E.G.; Berge, T.; Mero, I.L.; Berg-Hansen, P.; Aarseth, J.H.; Myhr, K.M.; Sondergaard, H.B.; Sellebjerg, F.; et al. Oligoclonal band phenotypes in MS differ in their HLA class II association, while specific KIR ligands at HLA class I show association to MS in general. J. Neuroimmunol. 2014, 274, 174–179. [Google Scholar] [CrossRef]
- Hollenbach, J.A.; Pando, M.J.; Caillier, S.J.; Gourraud, P.A.; Oksenberg, J.R. The killer immunoglobulin-like receptor KIR3DL1 in combination with HLA-Bw4 is protective against multiple sclerosis in African Americans. Genes Immun. 2016, 17, 199–202. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Discovery Dataset (ANZgene) | Replication Dataset (IMSGC + NBS) | ||
|---|---|---|---|
| Cases | Samples (n) | 1617 | 1313 |
| F | 1172 | 994 | |
| M | 445 | 319 | |
| F:M | 2.6 | 3.1 | |
| Controls | Samples (n) | 1988 | 1458 |
| F | 1231 | 753 | |
| M | 757 | 705 | |
| F:M | 1.6 | 1.1 | |
| Pre-imputed (# of SNPs) | HLA | 1047 | 62 |
| NKC | 137 | 33 | |
| LRC | 122 | 44 | |
| Total | 1306 | 139 | |
| Post-imputation overlapping (# of SNPs) | HLA | 2359 | |
| NKC | 2872 | ||
| LRC | 520 | ||
| Total | 5751 | ||
| Post-imputation ANZgene only (# of SNPs) | HLA | 54,541 | N/A |
| NKC | 3790 | ||
| LRC | 1576 | ||
| Total | 59,907 | ||
| HLA | NKC | LRC | ||||
|---|---|---|---|---|---|---|
| rsID | p Value | rsID | p Value | rid | p Value | |
| SNP | rs9271366 * | 1.83 × 10−61 | rs11053043 * | 9.91 × 10−4 | rs11672654 * | 3.29 × 10−3 |
| rs9267992 * | 1.30 × 10−59 | rs3764021 * | 1.08 × 10−3 | rs13344319 * | 0.0136 | |
| rs2395182 * | 7.59 × 10−50 | rs11052552 | 2.08 × 10−3 | rs4806741 * | 0.0153 | |
| rs3132946 | 1.83 × 10−48 | rs10844638 * | 2.27 × 10−3 | rs1671196 | 0.0269 | |
| rs3129941 * | 1.59 × 10−47 | rs10845080 * | 3.57 × 10−3 | rs10418607 * | 0.0270 | |
| Chromosome/Loci | rsID | Iterations (%) | p Value (Fisher’s Exact Testing) | Gene: Genetic Consequence |
|---|---|---|---|---|
| 6/HLA | rs2395182 * | 100 | 7.59 × 10−50 | HLA-DRA: 500B Downstream Variant |
| rs3117098 | 100 | 2.95 × 10−35 | HCG23: Non Coding Transcript Variant; LOC101929163: Intron Variant | |
| rs3129941 * | 100 | 1.59 × 10−47 | C6orf10: Missense Variant; LOC101929163: Intron Variant | |
| rs6903608 | 100 | 3.14 × 10−32 | INT(HLA-DRB9_HLA-DRB5) | |
| rs9271366 * | 100 | 1.83 × 10−61 | INT(HLA-DRB1_HLA-DQA1) | |
| rs9267992 * | 100 | 1.29 × 10−59 | INT(NOTCH4_TSPBP1-AS1) | |
| rs2854050 | 99.6 | 7.36 × 10−10 | NOTCH4: Intron Variant | |
| rs9277535 | 99.6 | 5.45 × 10−4 | HLA-DPB1: 3 Prime UTR Variant | |
| rs926070 | 99.2 | 2.74 × 10−36 | TSBP1-AS1: Intron Variant | |
| rs2281389 | 98.4 | 4.73 × 10−4 | HLA-DPA2: not reported | |
| rs2394160 | 98.4 | 3.00 × 10−12 | HLA-F: Intron Variant; HLA-F-AS1: Intron Variant | |
| rs2844482 | 98.1 | 0.673 | LTA: Intron Variant; LOC100287329: Intron Variant | |
| rs2647050 | 96.5 | 4.00 × 10−19 | INT(HLA-DQB1_MTC03P1) | |
| rs2856718 | 96.5 | 4.00 × 10−19 | INT(HLA-DQB1_MTC03P1) | |
| rs2395150 | 95.2 | 3.90 × 10−29 | C6orf10: Intron Variant; LOC101929163: Intron Variant | |
| rs1362126 | 94.3 | 7.31 × 10−13 | HLA-F: 2KB Upstream Variant | |
| rs3130299 | 94.3 | 7.53 × 10−11 | INT(NOTCH4_TSPBP1-AS1) | |
| rs2301271 | 94.1 | 2.35 × 10−23 | HLA-DQB2: Intron Variant | |
| rs1611285 | 93.6 | 6.58 × 10−6 | LOC105379663: Non Coding Transcript Variant | |
| rs7453920 | 92.9 | 2.40 × 10−23 | HLA-DQB2: Intron Variant | |
| rs2050190 | 92.7 | 2.43 × 10−9 | C6orf10: Intron Variant; LOC101929163: Intron Variant | |
| rs3819721 | 92.7 | 7.70 × 10−12 | TAP2: Intron Variant | |
| rs2284178 | 92.3 | 3.02 × 10−15 | HCP5: Non Coding Transcript Variant | |
| rs2051549 | 90.2 | 3.02 × 10−23 | HLA-DQB2: Intron Variant | |
| rs1077393 | 88.9 | 6.93 × 10−23 | BAG6: Intron Variant | |
| rs2647012 | 88.2 | 1.10 × 10−34 | INT(HLA-DQB1_MTC03P1) | |
| rs9275184 | 88.1 | 0.529 | INT(HLA-DQB1_MTC03P1) | |
| rs2395174 | 87 | 2.00 × 10−8 | INT(BTNL2_HLA-DRA) | |
| rs2394412 | 86.8 | 3.35 × 10−5 | LINC00243: Non Coding Transcript Variant | |
| rs2894046 | 86.8 | 3.35 × 10−5 | LINC00243: Non Coding Transcript Variant | |
| rs2975033 | 86.4 | 3.48 × 10−11 | LOC105375010: Intron Variant | |
| rs9277554 | 84.9 | 3.11 × 10−3 | HLA-DPB1: 3 Prime UTR Variant | |
| rs2848713 | 84.8 | 1.02 × 10−3 | INT(MICA_LINC01149) | |
| rs9296057 | 84.2 | 3.17 × 10−4 | LOC100294145: Non Coding Transcript Variant | |
| rs2517912 | 84 | 1.90 × 10−13 | INT(ZDHHC20P1_HLA-F) | |
| rs620202 | 83.4 | 2.00 × 10−7 | BRD2: Intron Variant | |
| rs2395349 | 82.8 | 2.37 × 10−5 | HLA-DPB2: Intron Variant | |
| rs2260000 | 82.7 | 1.25 × 10−19 | PRRC2A: Intron Variant | |
| rs6904029 | 82.6 | 3.75 × 10−11 | HCG9: Non Coding Transcript Variant | |
| rs2071653 | 82.3 | 1.10 × 10−11 | MOG: Intron Variant | |
| rs3117230 | 82.2 | 3.37 × 10−3 | INT(COL11A2PA1_HLA-DPB2) | |
| rs719653 | 81.4 | 5.24 × 10−26 | INT(HLA-DQB2_HLA-DOB) | |
| rs4151657 | 80.8 | 9.87 × 10−19 | CFB: Intron Variant | |
| rs2064478 | 79.2 | 3.69 × 10−3 | COL11A2PA1: not reported | |
| rs1035798 | 78.4 | 1.24 × 10−12 | AGER: Intron Variant | |
| rs3129882 | 78 | 3.58 × 10−23 | HLA-DRA: Intron Variant | |
| rs2395173 | 76.9 | 2.78 × 10−32 | INT(BTNL2_HLA-DRA) | |
| rs3135338 | 76 | 2.76 × 10−32 | INT(BTNL2_HLA-DRA) | |
| rs1611185 | 75.6 | 8.21 × 10−10 | HLA-P: not reported | |
| rs2299851 | 74.8 | 2.32 × 10−3 | MSH5: Intron Variant; MSH5-SAPCD1: Intron Variant | |
| rs1737046 | 74.2 | 2.77 × 10−10 | INT(LOC353010_HLA-V) | |
| rs6941112 | 72.2 | 2.51 × 10−18 | STK19: Intron Variant | |
| rs12665700 | 72 | 0.662 | MUC22: Missense Variant | |
| rs721394 | 71 | 0.417 | INT(HCG24_COL11A2) | |
| 12/NKC | rs10845080 * | 93.3 | 3.57 × 10−3 | KLRD1: Non Coding Transcript Variant |
| rs6488285 | 91.1 | 0.0259 | LOC101928100: Intron Variant | |
| rs3764021 * | 85.2 | 1.08 × 10−03 | CLEC2D: Synonymous Variant | |
| rs11053043 * | 82.7 | 9.91 × 10−4 | INT(CD69_KLRF1) | |
| rs10505741 | 79.9 | 0.0179 | CLEC2A: Intron Variant | |
| rs10844780 | 74.5 | 0.0103 | INT(CD69_KLRF1) | |
| rs10844638 * | 74 | 2.27 × 10−3 | INT(CLECL1_CD69) | |
| 19/LRC | rs11672654 * | 97.3 | 3.29 × 10−3 | LOC100421130 |
| rs6509868 | 82.5 | 0.0449 | INT(LAIR1_TTYH1) | |
| rs10411879 | 82 | 0.0706 | INT(LILRA1_LILRB1) | |
| rs4806741 * | 78.9 | 0.0153 | INT(LILRB2_LILRA5) | |
| rs11669029 | 77.6 | 0.0706 | INT(TARM1_OSCAR) | |
| rs10418607 * | 71.5 | 0.027 | INT(LILRA4_LAIR1) | |
| rs272411 | 70.9 | 0.0878 | LILRA1: Intron Variant | |
| rs13344319 * | 70.1 | 0.0136 | INT(LAIR1_TTYH1) | |
| rs2296371 | 70.1 | 0.185 | LILRP2: Non Coding Transcript Variant |
| CHR/Loci | Gene | Total # of SNPs | Lowest combined FDR Value | Genetic Consequence(s) |
|---|---|---|---|---|
| 6/HLA | BRD2 | 1 | 0.0767 | Intron Variant (1) |
| C2 | 4 | 3.18 × 10−5 | Intron Variant (4) | |
| CFB | 1 | 1.38 × 10−3 | Intron Variant (1) | |
| GPX5 | 2 | 6.86 × 10−3 | Intron Variant (1); Non-coding transcript variant (1) | |
| GPX6 | 3 | 6.86 × 10−3 | Intron Variant (2); missense variant (1) | |
| HLA-DOB | 3 | 0.0133 | Intron Variant (1); 2KB Upstream Variant (2) | |
| KIFC1 | 1 | 0.0693 | Intron Variant (1) | |
| LTA | 2 | 0.0129 | Downstream Variant (1); Missense Variant (1) | |
| LOC100287329 | 2 | 0.012855556 | 2KB Upstream Variant (2) | |
| PHF1 | 1 | 0.032153333 | Intron Variant (1) | |
| SYNGAP1 | 2 | 0.069256 | Intron Variant (2) | |
| TAP1 | 1 | 4.05E-03 | Intron Variant (1) | |
| TAP2 | 4 | 1.87 × 10−4 | Intron Variant (3); synonymous Variant (1) | |
| TNF | 1 | 0.012855556 | 2K Upstream Variant (1) | |
| TNXB | 10 | 1.11 × 10−4 | Intron Variant (9); Missense Variant (1) | |
| WDR46 | 1 | 0.0254 | Intron Variant (1) | |
| INT(FKBPL_PPT2) | 4 | 5.56 × 10−7 | NA/Intergenic | |
| INT(HLA-DOA_HLA-DPA1) | 2 | 0.0118 | ||
| INT(HLA-DQB2_HLA-DOB) | 4 | 4.05 × 10−3 | ||
| INT(PPP1R2P1_ LOC100294145) | 2 | 4.05 × 10−3 | ||
| INT(TAP1_PPP1R2P1) | 1 | 1.47 × 10−5 | ||
| INT(ZBTB9_BAK1) | 2 | 0.0235 | ||
| INT(ZSCAN23_GPX6) | 1 | 0.022343889 | ||
| 12/NKC | CLEC12B | 1 | 0.022577778 | Intron Variant (1) |
| LOC102724020 | 1 | 0.022577778 | Intron Variant (1) | |
| LOC112268091 | Intron Variant (1) | |||
| CLEC2A | 2 | 1.74 × 10−3 | Intron Variant (2) | |
| KLRF2 | 1 | 1.74 × 10−3 | Intron Variant (1) | |
| KLRA1P | 2 | 0.054753333 | Intron Variant (1); 2KB Upstream Variant | |
| KLRB1 | 1 | 0.093155556 | Intron Variant (1) | |
| KLRC4-KLRK1 readthrough | 1 | 0.083367778 | Intron Variant (1) | |
| KLRC4 | 1 | 0.083367778 | 2KB Upstream Variant (1) | |
| LINC02390 | 1 | 0.084108444 | Non-Coding Transcript Variant (1) | |
| LOC105369658 | 1 | 0.037733333 | Intron Variant (1) | |
| LOC374443, C-type lectin domain family 2 member D pseudogene | 2 | 0.012474 | Intron Variant (2) | |
| INT(KLRB1_CLEC2D) | 1 | 0.023585333 | NA/Intergenic | |
| INT(LINC02446_KLRA1P) | 2 | 0.0377 | ||
| INT(LOC408186_KLRB1) | 1 | 0.0286 | ||
| 19/LRC | RPS9 | 3 | 9.78 × 10−4 | Intron Variant (3) |
| INT(LILRA2_LILRB1) | 2 | 0.0520 | NA/Intergenic |
| GO (Biological Process) | ID | p-Value | q-Value FDR B & H | Hit Count in Query List | Hit Count in Genome | Hit in Query List |
|---|---|---|---|---|---|---|
| natural killer cell mediated cytotoxicity | GO:0042267 | 8.59 × 10−11 | 5.45 × 10−8 | 6 | 72 | KLRC4-KLRK1, TAP1, TAP2, KLRF2, CLEC12B, CLEC2A |
| natural killer cell mediated immunity | GO:0002228 | 1.11 × 10−10 | 5.45 × 10−8 | 6 | 75 | KLRC4-KLRK1, TAP1, TAP2, KLRF2, CLEC12B, CLEC2A |
| lymphocyte mediated immunity | GO:0002449 | 1.18 × 10−10 | 5.45 × 10−8 | 9 | 407 | C2, LTA, TNF, KLRC4-KLRK1, TAP1, TAP2, KLRF2, CLEC12B, CLEC2A |
| leukocyte mediated cytotoxicity | GO:0001909 | 3.63 × 10−9 | 1.25 × 10−6 | 6 | 133 | KLRC4-KLRK1, TAP1, TAP2, KLRF2, CLEC12B, CLEC2A |
| regulation of lymphocyte mediated immunity | GO:0002706 | 2.54 × 10−8 | 7.03 × 10−6 | 6 | 184 | LTA, TNF, KLRC4-KLRK1, TAP1, TAP2, CLEC12B |
| regulation of immune effector process | GO:0002697 | 3.06 × 10−8 | 7.05 × 10−6 | 8 | 527 | C2, LTA, TNF, KLRC4-KLRK1, TAP1, TAP2, CFB, CLEC12B |
| cell killing | GO:0001906 | 6.96 × 10−8 | 1.38 × 10−5 | 6 | 218 | KLRC4-KLRK1, TAP1, TAP2, KLRF2, CLEC12B, CLEC2A |
| GO (Molecular Function) | ||||||
| tapasin binding | GO:0046980 | 1.12 × 10−6 | 1.36 × 10−4 | 2 | 2 | TAP1, TAP2 |
| ABC-type peptide transporter activity | GO:0015440 | 6.68 × 10−6 | 2.04 × 10−4 | 2 | 4 | TAP1, TAP2 |
| ABC-type peptide antigen transporter activity | GO:0015433 | 6.68 × 10−6 | 2.04 × 10−4 | 2 | 4 | TAP1, TAP2 |
| TAP2 binding | GO:0046979 | 6.68 × 10−6 | 2.04 × 10−4 | 2 | 4 | TAP1, TAP2 |
| TAP1 binding | GO:0046978 | 1.11 × 10−5 | 2.65 × 10−4 | 2 | 5 | TAP1, TAP2 |
| carbohydrate binding | GO:0030246 | 1.31 × 10−5 | 2.65 × 10−4 | 5 | 295 | KLRC4-KLRK1, KLRB1, KLRF2, CLEC12B, CLEC2A |
| TAP binding | GO:0046977 | 3.11 × 10−5 | 5.42 × 10−4 | 2 | 8 | TAP1, TAP2 |
| MHC protein binding | GO:0042287 | 4.15 × 10−5 | 6.34 × 10−4 | 3 | 63 | HLA-DOB, TAP1, TAP2 |
| MHC class Ib protein binding | GO:0023029 | 8.63 × 10−5 | 1.17 × 10−3 | 2 | 13 | TAP1, TAP2 |
| ABC-type transporter activity | GO:0140359 | 2.54 × 10−4 | 2.82 × 10−3 | 2 | 22 | TAP1, TAP2 |
| glutathione peroxidase activity | GO:0004602 | 2.54 × 10−4 | 2.82 × 10−3 | 2 | 22 | GPX5, GPX6 |
| GO (Pathway) | ||||||
| Antigen processing and presentation | 83074 (KEGG) | 9.58 × 10−8 | 2.08 × 10−5 | 5 | 77 | TNF, HLA-DOB, TAP1, TAP2, KLRC4 |
| Herpes simplex infection | 377873 (KEGG) | 7.45 × 10−6 | 8.09 × 10−4 | 5 | 185 | LTA, TNF, HLA-DOB, TAP1, TAP2 |
| Type I diabetes mellitus | 83095 (Reactome) | 3.74 × 10−5 | 2.41 × 10−3 | 3 | 43 | LTA, TNF, HLA-DOB |
| Activation of C3 and C5 | 1269248 (KEGG) | 4.73 × 10−5 | 2.41 × 10−3 | 2 | 7 | C2, CFB |
| Malaria | 152665 (KEGG) | 5.55 × 10−5 | 2.41 × 10−3 | 3 | 49 | TNF, KLRC4-KLRK1, KLRB1 |
| Staphylococcus aureus infection | 172846 (KEGG) | 8.30 × 10−5 | 3.00 × 10−3 | 3 | 56 | C2, HLA-DOB, CFB |
| Antigen Presentation: Folding, assembly and peptide loading of class I MHC | 1269194 (Reactome) | 6.64 × 10−4 | 2.06 × 10−2 | 2 | 25 | TAP1, TAP2 |
| Regulation of Complement cascade | 1269250 (Reactome) | 7.76 × 10−4 | 2.10 × 10−2 | 2 | 27 | C2, CFB |
| Asthma | 83120 (KEGG) | 1.02 × 10−3 | 2.10 × 10−2 | 2 | 31 | TNF, HLA-DOB |
| Initial triggering of complement | 1269242 (Reactome) | 1.02 × 10−3 | 2.10 × 10−2 | 2 | 31 | C2, CFB |
| Systemic lupus erythematosus | 83122 (KEGG) | 1.06 × 10−3 | 2.10 × 10−2 | 3 | 133 | C2, TNF, HLA-DOB |
| Primary immunodeficiency | 83125 (KEGG) | 1.46 × 10−3 | 2.35 × 10−2 | 2 | 37 | TAP1, TAP2 |
| Detoxification of Reactive Oxygen Species | 1270420 (Reactome) | 1.54 × 10−3 | 2.35 × 10−2 | 2 | 38 | GPX5, GPX6 |
| SNP1 | SNP2 | Epistasis Interaction | |||
|---|---|---|---|---|---|
| Chr:bp | Genomic Location | Chr:bp | Genomic Location | OR_INT | P |
| 6:32829320 | INT(TAP1_PPP1R2P1) | 12:10750157 | KLRA1P | 0.48 | 0.000255 |
| 6:32832786 | INT(TAP1_PPP1R2P1) | 12:10750157 | KLRA1P | 0.487 | 0.000685 |
| 6:32102305 | INT(FKBPL_PPT2) | 19:55116651 | INT(LILRA1_LILRB1) | 0.632 | 0.00306 |
| 6:32112626 | INT(FKBPL_PPT2) | 19:55116651 | INT(LILRA1_LILRB1) | 0.639 | 0.00392 |
| 6:32069806 | TNXB | 19:55116651 | INT(LILRA1_LILRB1) | 0.636 | 0.00418 |
| 6:31916400 | CFB | 12:9742327 | INT(LOC408186_KLRB1) | 1.76 | 0.00526 |
| 6:32109165 | INT(FKBPL_PPT2) | 19:55116651 | INT(LILRA1_LILRB1) | 0.646 | 0.00561 |
| 6:32026257 | TNXB | 19:55116651 | INT(LILRA1_LILRB1) | 0.648 | 0.00601 |
| 6:31879158 | C2 | 19:55116651 | INT(LILRA1_LILRB1) | 0.662 | 0.0105 |
| 6:31888367 | C2 | 19:55116651 | INT(LILRA1_LILRB1) | 0.667 | 0.012 |
| 6:31884823 | C2 | 19:55116651 | INT(LILRA1_LILRB1) | 0.672 | 0.0137 |
| 6:32852448 | INT(PPP1R2P1_LOC100294145) | 12:9742327 | INT(LOC408186_KLRB1) | 2.8 | 0.0162 |
| 6:31542308 | TNF; LTA; LOC100287329 | 12:9742327 | INT(LOC408186_KLRB1) | 0.561 | 0.018 |
| 6:32069806 | TNXB | 12:10750157 | KLRA1P | 0.679 | 0.022 |
| 6:32026257 | TNXB | 12:10750157 | KLRA1P | 0.682 | 0.0239 |
| 6:32112626 | INT(FKBPL_PPT2) | 12:10044542 | CLEC2A; KLRF2 | 6.42 | 0.0287 |
| 6:32109165 | INT(FKBPL_PPT2) | 12:10044542 | CLEC2A; KLRF2 | 6.23 | 0.0314 |
| 6:32938199 | BRD2 | 12:10169041 | CLEC12B; LOC102724020; LOC112268091 | 0.722 | 0.0423 |
| 6:32057972 | TNXB | 12:10700014 | LOC105369658 | 0.142 | 0.0432 |
| 6:32010272 | TNXB | 12:10700014 | LOC105369658 | 0.142 | 0.0434 |
| 6:32021838 | TNXB | 12:10700014 | LOC105369658 | 0.142 | 0.0434 |
| 6:32030284 | TNXB | 12:10700014 | LOC105369658 | 0.142 | 0.0435 |
| 6:32019746 | TNXB | 12:10700014 | LOC105369658 | 0.142 | 0.0436 |
| 6:31540556 | LTA; LOC100287329 | 12:9742327 | INT(LOC408186_KLRB1) | 0.629 | 0.0467 |
| 6:31916400 | CFB | 19:55116651 | INT(LILRA1_LILRB1) | 0.785 | 0.0472 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burnard, S.M.; Lea, R.A.; Benton, M.; Eccles, D.; Kennedy, D.W.; Lechner-Scott, J.; Scott, R.J. Capturing SNP Association across the NK Receptor and HLA Gene Regions in Multiple Sclerosis by Targeted Penalised Regression Models. Genes 2022, 13, 87. https://doi.org/10.3390/genes13010087
Burnard SM, Lea RA, Benton M, Eccles D, Kennedy DW, Lechner-Scott J, Scott RJ. Capturing SNP Association across the NK Receptor and HLA Gene Regions in Multiple Sclerosis by Targeted Penalised Regression Models. Genes. 2022; 13(1):87. https://doi.org/10.3390/genes13010087
Chicago/Turabian StyleBurnard, Sean M., Rodney A. Lea, Miles Benton, David Eccles, Daniel W. Kennedy, Jeannette Lechner-Scott, and Rodney J. Scott. 2022. "Capturing SNP Association across the NK Receptor and HLA Gene Regions in Multiple Sclerosis by Targeted Penalised Regression Models" Genes 13, no. 1: 87. https://doi.org/10.3390/genes13010087
APA StyleBurnard, S. M., Lea, R. A., Benton, M., Eccles, D., Kennedy, D. W., Lechner-Scott, J., & Scott, R. J. (2022). Capturing SNP Association across the NK Receptor and HLA Gene Regions in Multiple Sclerosis by Targeted Penalised Regression Models. Genes, 13(1), 87. https://doi.org/10.3390/genes13010087