

Understanding the Roles of the NSD Protein Methyltransferases in Head and Neck Squamous Cell Carcinoma


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Structure of NSD Family of Protein Methyltransferases
3.2. Substrates of NSD Protein Methyltransferases
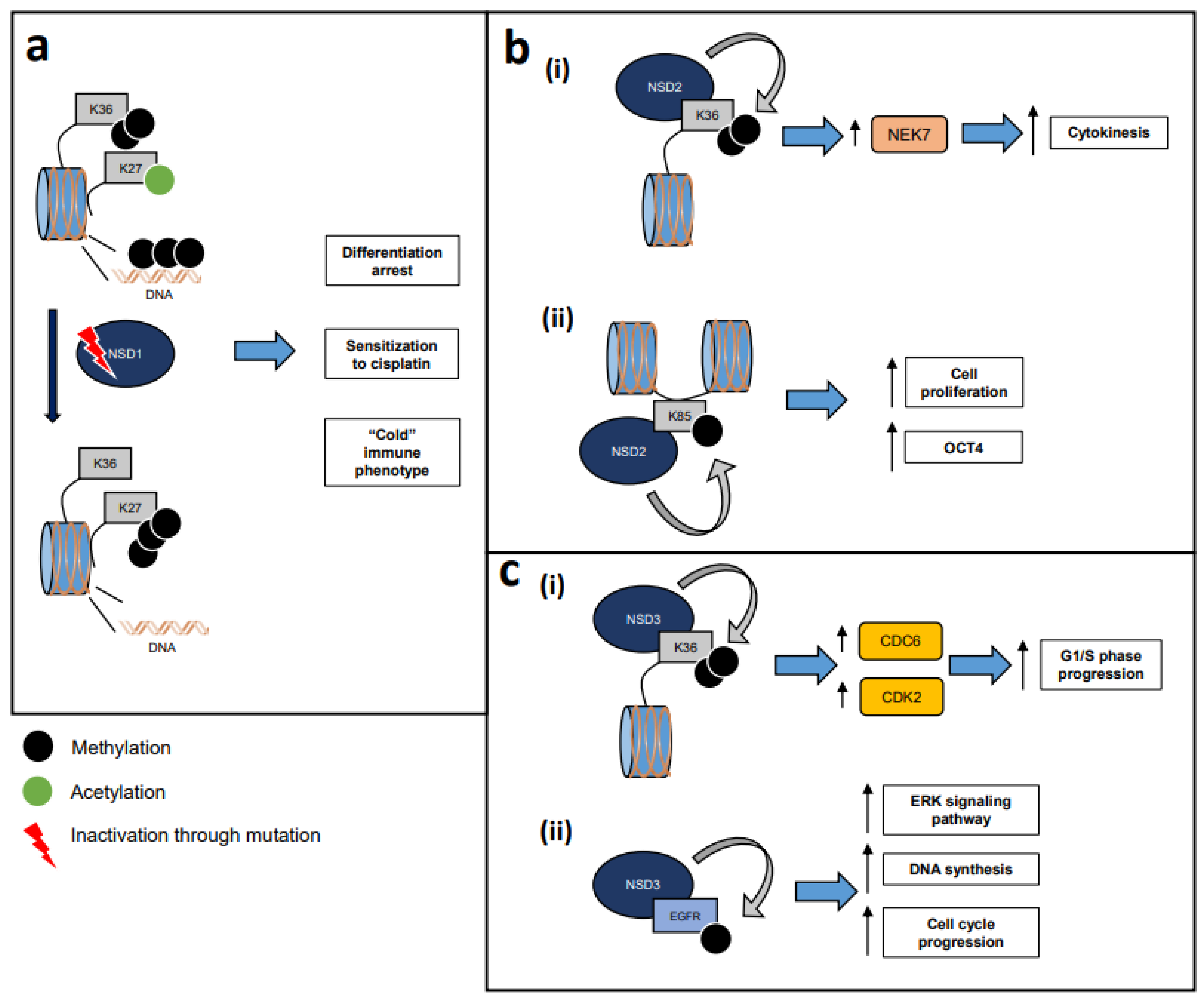
3.3. Physiological Functions of NSD1, NSD2, and NSD3
3.4. Pathobiological Implications of the NSDs in HNSCC
3.4.1. The NSDs in HPV-Negative HNSCC
3.4.2. NSD1 in HPV-Negative HNSCC
3.4.3. NSD2 in HPV-Negative HNSCC
3.4.4. NSD3 in HPV-Negative HNSCC
3.4.5. The NSDs in HPV-Positive HNSCC
3.5. Clinical/Translational Implications of the NSDs in HNSCC
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Sturgis, E.M. Human Papillomavirus as a Marker of the Natural History and Response to Therapy of Head and Neck Squamous Cell Carcinoma. Semin. Radiat. Oncol. 2012, 22, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human Papillomavirus and Rising Oropharyngeal Cancer Incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Saloura, V.; Vougiouklakis, T.; Sievers, C.; Burkitt, K.; Nakamura, Y.; Hager, G.; van Waes, C. The role of protein methyltransferases as potential novel therapeutic targets in squamous cell carcinoma of the head and neck. Oral Oncol. 2018, 81, 100–108, Erratum in Oral Oncol. 2018, 86, 318. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kee, H.J.; Choe, N.W.; Kim, S.M.; Eom, G.H.; Baek, H.J.; Kook, H.; Seo, S.B. Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol. Cell. Biol. 2008, 28, 2023–2034. [Google Scholar] [CrossRef] [Green Version]
- Marango, J.; Shimoyama, M.; Nishio, H.; Meyer, J.A.; Min, D.-J.; Sirulnik, A.; Martinez-Martinez, Y.; Chesi, M.; Bergsagel, P.L.; Zhou, M.-M.; et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 2008, 111, 3145–3154. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Trojer, P.; Xu, C.-F.; Cheung, P.; Kuo, A.; Drury, W.J.; Qiao, Q.; Neubert, T.A.; Xu, R.-M.; Gozani, O.; et al. The Target of the NSD Family of Histone Lysine Methyltransferases Depends on the Nature of the Substrate. J. Biol. Chem. 2009, 284, 34283–34295. [Google Scholar] [CrossRef]
- Nimura, K.; Ura, K.; Shiratori, H.; Ikawa, M.; Okabe, M.; Schwartz, R.J.; Kaneda, Y. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf–Hirschhorn syndrome. Nature 2009, 460, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Lucio-Eterovic, A.K.; Singh, M.M.; Gardner, J.E.; Veerappan, C.S.; Rice, J.C.; Carpenter, P.B. Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc. Natl. Acad. Sci. USA 2010, 107, 16952–16957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, A.J.; Cheung, P.; Chen, K.; Zee, B.M.; Kioi, M.; Lauring, J.; Xi, Y.; Park, B.H.; Shi, X.; Garcia, B.A.; et al. NSD2 links demethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 2011, 44, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Q.; Li, Y.; Chen, Z.; Wang, M.; Reinberg, D.; Xu, R.M. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J. Biol. Chem. 2011, 286, 8361–8368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 Extraterminal Domain Confers Transcription Activation Independent of pTEFb by Recruiting Multiple Proteins, Including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [Green Version]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and Comparative Genomic Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.; Swaroop, A.; Troche, C.; Licht, J.D. The Role of Nuclear Receptor–Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026708. [Google Scholar] [CrossRef] [Green Version]
- Vougiouklakis, T.; Hamamoto, R.; Nakamura, Y.; Saloura, V. The NSD family of protein methyltransferases in human cancer. Epigenomics 2015, 7, 863–874. [Google Scholar] [CrossRef]
- Jaju, R.J.; Fidler, C.; Haas, O.A.; Strickson, A.J.; Watkins, F.; Clark, K.; Cross, N.C.; Cheng, J.F.; Aplan, P.D.; Kearney, L.; et al. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood 2001, 98, 1264–1267. [Google Scholar] [CrossRef] [Green Version]
- Shiba, N.; Ichikawa, H.; Taki, T.; Park, M.J.; Jo, A.; Mitani, S.; Kobayashi, T.; Shimada, A.; Sotomatsu, M.; Arakawa, H.; et al. NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer 2013, 52, 683–693. [Google Scholar]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Stec, I.; Wright, T.J.; van Ommen, G.J.B.; de Boer, P.A.; van Haeringen, A.; Moorman, A.F.; Altherr, M.R.; den Dunnen, J.T. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf–Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum. Mol. Genet. 1998, 7, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Nardini, E.; Lim, R.S.; Smith, K.D.; Kuehl, W.M.; Bergsagel, P.L. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 1998, 92, 3025–3034. [Google Scholar] [CrossRef] [PubMed]
- Maiolo, A.T.; Neri, A.; Finelli, P.; Fabris, S.; Zagano, S.; Baldini, L.; Intini, D.; Nobili, L.; Lombardi, L. Detection of t(4;14)(p16.3;q32) chromosomal translocation in multiple myeloma by double-color fluorescent in situ hybridization. Blood 1999, 94, 724–732. [Google Scholar]
- Lauring, J.; Abukhdeir, A.M.; Konishi, H.; Garay, J.P.; Gustin, J.P.; Wang, Q.; Arceci, R.J.; Matsui, W.; Park, B.H. The multiple myeloma–associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 2008, 111, 856–864. [Google Scholar] [CrossRef]
- Brito, J.L.; Walker, B.; Jenner, M.; Dickens, N.J.; Brown, N.J.; Ross, F.M.; Avramidou, A.; Irving, J.A.; Gonzalez, D.; Davies, F.E.; et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica 2009, 94, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Garcia, E.; Popovic, R.; Min, D.-J.; Sweet, S.; Thomas, P.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, H.; Chuai, S.; Xu, F.; Yan, F.; Englund, N.; Wang, Z.; Zhang, H.; Fang, M.; Wang, Y.; et al. NSD2 Is Recruited through Its PHD Domain to Oncogenic Gene Loci to Drive Multiple Myeloma. Cancer Res. 2013, 73, 6277–6288. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.Y.; Martinez-Garcia, E.; Phillip, J.M.; Chambliss, A.B.; Popovic, R.; Ezponda, T.; Small, E.C.; Will, C.; Phillip, M.P.; Neri, P.; et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016, 35, 5905–5915. [Google Scholar] [CrossRef]
- Zheng, Y.; Sweet, S.M.M.; Popovic, R.; Martinez-Garcia, E.; Tipton, J.D.; Thomas, P.M.; Licht, J.D.; Kelleher, N.L. Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc. Natl. Acad. Sci. USA 2012, 109, 13549–13554. [Google Scholar] [CrossRef] [Green Version]
- Angrand, P.-O.; Apiou, F.; Stewart, A.; Dutrillaux, B.; Losson, R.; Chambon, P. NSD3, a New SET Domain-Containing Gene, Maps to 8p12 and Is Amplified in Human Breast Cancer Cell Lines. Genomics 2001, 74, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Stec, I.; van Ommen, G.-J.B.; den Dunnen, J.T. WHSC1L1, on Human Chromosome 8p11.2, Closely Resembles WHSC1 and Maps to a Duplicated Region Shared with 4p16.3. Genomics 2001, 76, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McGee, J.; Chen, X.; Doman, T.N.; Gong, X.; Zhang, Y.; Hamm, N.; Ma, X.; Higgs, R.E.; Bhagwat, S.V.; et al. Identification of Druggable Cancer Driver Genes Amplified across TCGA Datasets. PLoS ONE 2014, 9, e98293. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Q.; Liu, G.; Bollig-Fischer, A.; Giroux, C.N.; Ethier, S.P. Transforming Properties of 8p11-12 Amplified Genes in Human Breast Cancer. Cancer Res. 2010, 70, 8487–8497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Thomsen, R.; Kahns, S.; Nielsen, A.L. The NSD3L histone methyltransferase regulates cell cycle and cell invasion in breast cancer cells. Biochem. Biophys. Res. Commun. 2010, 398, 565–570. [Google Scholar] [CrossRef]
- Kang, D.; Cho, H.S.; Toyokawa, G.; Kogure, M.; Yamane, Y.; Iwai, Y.; Hayami, S.; Tsunoda, T.; Field, H.I.; Matsuda, K.; et al. The histone methyltransferase Wolf–Hirschhorn syndrome candidate 1-like 1 (WHSC1L1) is involved in human carcinogenesis. Genes Chromosomes Cancer 2013, 52, 126–139. [Google Scholar] [CrossRef]
- Yuan, G.; Flores, N.M.; Hausmann, S.; Lofgren, S.M.; Kharchenko, V.; Angulo-Ibanez, M.; Sengupta, D.; Lu, X.; Czaban, I.; Azhibek, D.; et al. Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature 2021, 590, 504–508. [Google Scholar] [CrossRef]
- Baker, L.A.; Allis, C.D.; Wang, G.G. PHD fingers in human diseases: Disorders arising from misinterpreting epigenetic marks. Mutat. Res. Mol. Mech. Mutagen. 2008, 647, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Pasillas, M.P.; Shah, M.; Kamps, M.P. NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Hum. Mutat. 2010, 32, 292–298. [Google Scholar] [CrossRef]
- Sankaran, S.M.; Wilkinson, A.W.; Elias, J.E.; Gozani, O. A PWWP Domain of Histone-Lysine N-Methyltransferase NSD2 Binds to Dimethylated Lys-36 of Histone H3 and Regulates NSD2 Function at Chromatin. J. Biol. Chem. 2016, 291, 8465–8474. [Google Scholar] [CrossRef] [Green Version]
- Morishita, M.; di Luccio, E. Cancers and the NSD family of histone lysine methyltransferases. Biochim. Biophys. Acta 2011, 1816, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The SET-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005, 6, 227. [Google Scholar] [CrossRef] [PubMed]
- Herz, H.-M.; Garruss, A.; Shilatifard, A. SET for life: Biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013, 38, 621–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.; Baur, E.V.; Garnier, J.; Lerouge, T.; Vonesch, J.; Lutz, Y.; Chambon, P.; Losson, R. Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J. 1998, 17, 3398–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, B.; Shibata, Y.; Strahl, B.D.; Lieb, J.D. Dimethylation of Histone H3 at Lysine 36 DemarcatesRegulatory and Nonregulatory ChromatinGenome-Wide. Mol. Cell. Biol. 2005, 25, 9447–9459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Tian, W.; Yuan, G.; Deng, P.; Sengupta, D.; Cheng, Z.; Cao, Y.; Ren, J.; Qin, Y.; Zhou, Y.; et al. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature 2020, 590, 498–503. [Google Scholar] [CrossRef]
- Saloura, V.; Vougiouklakis, T.; Bao, R.; Kim, S.; Baek, S.; Zewde, M.; Bernard, B.; Burkitt, K.; Nigam, N.; Izumchenko, E.; et al. WHSC1 monomethylates histone H1 and induces stem-cell like features in squamous cell carcinoma of the head and neck. Neoplasia 2020, 22, 283–293. [Google Scholar] [CrossRef]
- Lu, T.; Jackson, M.W.; Wang, B.; Yang, M.; Chance, M.R.; Miyagi, M.; Gudkov, A.V.; Stark, G.R. Regulation of NF-κB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. USA 2009, 107, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lee, Y.-R.; Dang, F.; Gan, W.; Menon, A.V.; Katon, J.M.; Hsu, C.-H.; Asara, J.M.; Tibarewal, P.; Leslie, N.R.; et al. PTEN Methylation by NSD2 Controls Cellular Sensitivity to DNA Damage. Cancer Discov. 2019, 9, 1306–1323. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, Q.; Xu, X.; Xie, B.; Zhao, Y.; Li, N.; Cao, X. The methyltransferase NSD3 promotes antiviral innate immunity via direct lysine methylation of IRF3. J. Exp. Med. 2017, 214, 3597–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saloura, V.; Vougiouklakis, T.; Zewde, M.; Deng, X.; Kiyotani, K.; Park, J.-H.; Matsuo, Y.; Lingen, M.; Suzuki, T.; Dohmae, N.; et al. WHSC1L1-mediated EGFR mono-methylation enhances the cytoplasmic and nuclear oncogenic activity of EGFR in head and neck cancer. Sci. Rep. 2017, 7, 40664. [Google Scholar] [CrossRef] [PubMed]
- Rayasam, G.V.; Wendling, O.; Angrand, P.-O.; Mark, M.; Niederreither, K.; Song, L.; Lerouge, T.; Hager, G.L.; Chambon, P.; Losson, R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003, 22, 3153–3163. [Google Scholar] [CrossRef] [Green Version]
- Kurotaki, N.; Imaizumi, K.; Harada, N.; Masuno, M.; Kondoh, T.; Nagai, T.; Ohashi, H.; Naritomi, K.; Tsukahara, M.; Makita, Y.; et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 2002, 30, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Leventopoulos, G.; Kitsiou-Tzeli, S.; Kritikos, K.; Psoni, S.; Mavrou, A.; Kanavakis, E.; Fryssira, H. A Clinical Study of Sotos Syndrome Patients With Review of the Literature. Pediatr. Neurol. 2009, 40, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Schellmoser, S.; Kraus, C.; Dorr, H.G.; Trautmann, U.; Altherr, M.R.; Pfeiffer, R.A.; Reis, A. First known microdeletion within the Wolf-Hirschhorn syndrome critical region refines genotype-phenotype correlation. Am. J. Med. Genet. 2001, 99, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017, 49, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Izreig, S.; Yarbrough, W.G.; Issaeva, N. NSD1 mutations by HPV status in head and neck cancer: Differences in survival and response to DNA-damaging agents. Cancers Head Neck 2019, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Peri, S.; Izumchenko, E.; Schubert, A.D.; Slifker, M.J.; Ruth, K.; Serebriiskii, I.G.; Guo, T.; Burtness, B.A.; Mehra, R.; Ross, E.A.; et al. NSD1- and NSD2-damaging mutations define a subset of laryngeal tumors with favorable prognosis. Nat. Commun. 2017, 8, 1772. [Google Scholar] [CrossRef] [Green Version]
- Bui, N.; Huang, J.K.; Bojorquez-Gomez, A.; Licon, K.; Sanchez, K.S.; Tang, S.N.; Beckett, A.N.; Wang, T.; Zhang, W.; Shen, J.P.; et al. Disruption of NSD1 in Head and Neck Cancer Promotes Favorable Chemotherapeutic Responses Linked to Hypomethylation. Mol. Cancer Ther. 2018, 17, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- Farhangdoost, N.; Horth, C.; Hu, B.; Bareke, E.; Chen, X.; Li, Y.; Coradin, M.; Garcia, B.A.; Lu, C.; Majewski, J. Chromatin dysregulation associated with NSD1 mutation in head and neck squamous cell carcinoma. Cell Rep. 2021, 34, 108769. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Shin, J.H.; Tay, J.K.; Prunello, M.; Gentles, A.J.; Sunwoo, J.B.; Gevaert, O. NSD1 inactivation defines an immune cold, DNA hypomethylated subtype in squamous cell carcinoma. Sci. Rep. 2017, 7, 17064. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Goldberg, E.M.; Chen, X.; Xu, X.; McGuire, J.T.; Leuzzi, G.; Karagiannis, D.; Tate, T.; Farhangdoost, N.; Horth, C.; et al. Histone methylation antagonism drives tumor immune evasion in squamous cell carcinomas. Mol. Cell 2022, 82, 3901–3918. [Google Scholar] [CrossRef] [PubMed]
- Saloura, V.; Cho, H.-S.; Kiyotani, K.; Alachkar, H.; Zuo, Z.; Nakakido, M.; Tsunoda, T.; Seiwert, T.; Lingen, M.; Licht, J.; et al. WHSC1 Promotes Oncogenesis through Regulation of NIMA-Related Kinase-7 in Squamous Cell Carcinoma of the Head and Neck. Mol. Cancer Res. 2015, 13, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Saloura, V.; Vougiouklakis, T.; Zewde, M.; Kiyotani, K.; Park, J.-H.; Gao, G.; Karrison, T.; Lingen, M.; Nakamura, Y.; Hamamoto, R. WHSC1L1 drives cell cycle progression through transcriptional regulation of CDC6 and CDK2 in squamous cell carcinoma of the head and neck. Oncotarget 2016, 7, 42527–42538. [Google Scholar] [CrossRef]
- Xue, W.; Shen, Z.; Li, L.; Zheng, Y.; Yan, D.; Kan, Q.; Zhao, J. Long non-coding RNAs MACC1-AS1 and FOXD2-AS1 mediate NSD2-induced cisplatin resistance in esophageal squamous cell carcinoma. Mol. Ther. Nucleic Acids 2020, 23, 592–602. [Google Scholar] [CrossRef]
- French, C.A. NUT Carcinoma: Clinicopathologic features, pathogenesis, and treatment. Pathol. Int. 2018, 68, 583–595. [Google Scholar] [CrossRef]
- French, C.A.; Rahman, S.; Walsh, E.M.; Kühnle, S.; Grayson, A.R.; Lemieux, M.E.; Grunfeld, N.; Rubin, B.P.; Antonescu, C.R.; Zhang, S.; et al. NSD3–NUT Fusion Oncoprotein in NUT Midline Carcinoma: Implications for a Novel Oncogenic Mechanism. Cancer Discov. 2014, 4, 928–941. [Google Scholar] [CrossRef] [Green Version]
- Haft, S.; Ren, S.; Xu, G.; Mark, A.; Fisch, K.; Guo, T.W.; Khan, Z.; Pang, J.; Ando, M.; Liu, C.; et al. Mutation of chromatin regulators and focal hotspot alterations characterize human papillomavirus–positive oropharyngeal squamous cell carcinoma. Cancer 2019, 125, 2423–2434. [Google Scholar] [CrossRef]
- Gameiro, S.F.; Ghasemi, F.; Zeng, P.Y.F.; Mundi, N.; Howlett, C.J.; Plantinga, P.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. Low expression of NSD1, NSD2, and NSD3 define a subset of human papillomavirus-positive oral squamous carcinomas with unfavorable prognosis. Infect. Agents Cancer 2021, 16, 13. [Google Scholar] [CrossRef]
- Allali-Hassani, A.; Kuznetsova, E.; Hajian, T.; Wu, H.; Dombrovski, L.; Li, Y.; Gräslund, S.; Arrowsmith, C.H.; Schapira, M.; Vedadi, M. A Basic Post-SET Extension of NSDs Is Essential for Nucleosome Binding In Vitro. SLAS Discov. Adv. Sci. Drug Discov. 2014, 19, 928–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilworth, D.; Hanley, R.P.; de Freitas, R.F.; Allali-Hassani, A.; Zhou, M.; Mehta, N.; Marunde, M.R.; Ackloo, S.; Machado, R.A.C.; Yazdi, A.K.; et al. A chemical probe targeting the PWWP domain alters NSD2 nucleolar localization. Nat. Chem. Biol. 2021, 18, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, J.K.; Iwata, K.; Ooike, S.; Sugahara, K.; Nakamura, H.; Daibata, M. Development of the BET bromodomain inhibitor OTX015. Mol. Cancer Ther. 2013, 12 (Suppl. 11), C244. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Côté, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Rhyasen, G.W.; Hattersley, M.M.; Yao, Y.; Dulak, A.; Wang, W.; Petteruti, P.; Dale, I.L.; Boiko, S.; Cheung, T.; Zhang, J.; et al. AZD5153: A Novel Bivalent BET Bromodomain Inhibitor Highly Active against Hematologic Malignancies. Mol. Cancer Ther. 2016, 15, 2563–2574. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.R.; Schweizer, M.T.; Nanus, D.M.; Pantuck, A.J.; Heath, E.I.; Campeau, E.; Attwell, S.; Norek, K.; Snyder, M.; Bauman, L.; et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in Combination with Enzalutamide in Patients with Metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. 2020, 26, 5338–5347. [Google Scholar] [CrossRef]


{kind=link}
| NSD Type | Substrate | Enzyme Activity | References |
|---|---|---|---|
| Histone substrates | |||
| NSD1, 2, 3 | H3K36 | Mono- or di-methylation of H3K36 | [9,10,11,12,13] |
| NSD2 | H1K85 | Mono-methylation of H1K85 | [48] |
| Non-histone substrates | |||
| NSD1 | NF-κB | Methylation of K218 and K221 of the RELA/p65 subunit of NF-κB | [49] |
| NSD2 | PTEN | Methylation of PTEN at K349 | [50] |
| NSD3 | IRF3 | Methylation of IRF3 through PWWP1 domain at K366 | [51] |
| NSD3 | EGFR | Methylation of EGFR at K721 | [52] |
| NSD Type | Function | References |
|---|---|---|
| NSD1 |
| [57,58,59,60,61,62,63] |
| NSD2 |
| [48,64] |
| NSD3 |
| [52,65] |
| NSD Type | Inhibitor | Mechanism of Action | References |
|---|---|---|---|
| Non-bromodomain inhibitors | |||
| NSD1, 2, 3 | Suramin | Nonspecific inhibitor of DNA-binding proteins | [71] |
| NSD2 | UNC6934 | Targets the PWWP1 domain of NSD2, disrupting its interaction with H3K36me2 nucleosomes | [72] |
| Bromodomain inhibitors | |||
| NSD3 | (+)-JQ1 | Binds to all bromodomains within the BET family | [73] |
| NSD3 | Birabresib (OTX015) | Bromodomain inhibitor for BRD2, BRD3, and BRD4 | [74] |
| NSD3 | Pelabresib (CPI-0610) | Selective benzoisoxaoloazepine BET bromodomain inhibitor for BRD4-BD1 | [75] |
| NSD3 NSD3 | AZD 5153 6-hydroxy 2-naphthoic acid ZEN-3694 | Orally available BET-BRD4 bromodomain inhibitor Orally available pan-BET bromodomain inhibitor | [76] [77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murali, M.; Saloura, V. Understanding the Roles of the NSD Protein Methyltransferases in Head and Neck Squamous Cell Carcinoma. Genes 2022, 13, 2013. https://doi.org/10.3390/genes13112013
Murali M, Saloura V. Understanding the Roles of the NSD Protein Methyltransferases in Head and Neck Squamous Cell Carcinoma. Genes. 2022; 13(11):2013. https://doi.org/10.3390/genes13112013
Chicago/Turabian StyleMurali, Madhavi, and Vassiliki Saloura. 2022. "Understanding the Roles of the NSD Protein Methyltransferases in Head and Neck Squamous Cell Carcinoma" Genes 13, no. 11: 2013. https://doi.org/10.3390/genes13112013
APA StyleMurali, M., & Saloura, V. (2022). Understanding the Roles of the NSD Protein Methyltransferases in Head and Neck Squamous Cell Carcinoma. Genes, 13(11), 2013. https://doi.org/10.3390/genes13112013