
Implementation of Public Funded Genome Sequencing in Evaluation of Fetal Structural Anomalies

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Material and Methods
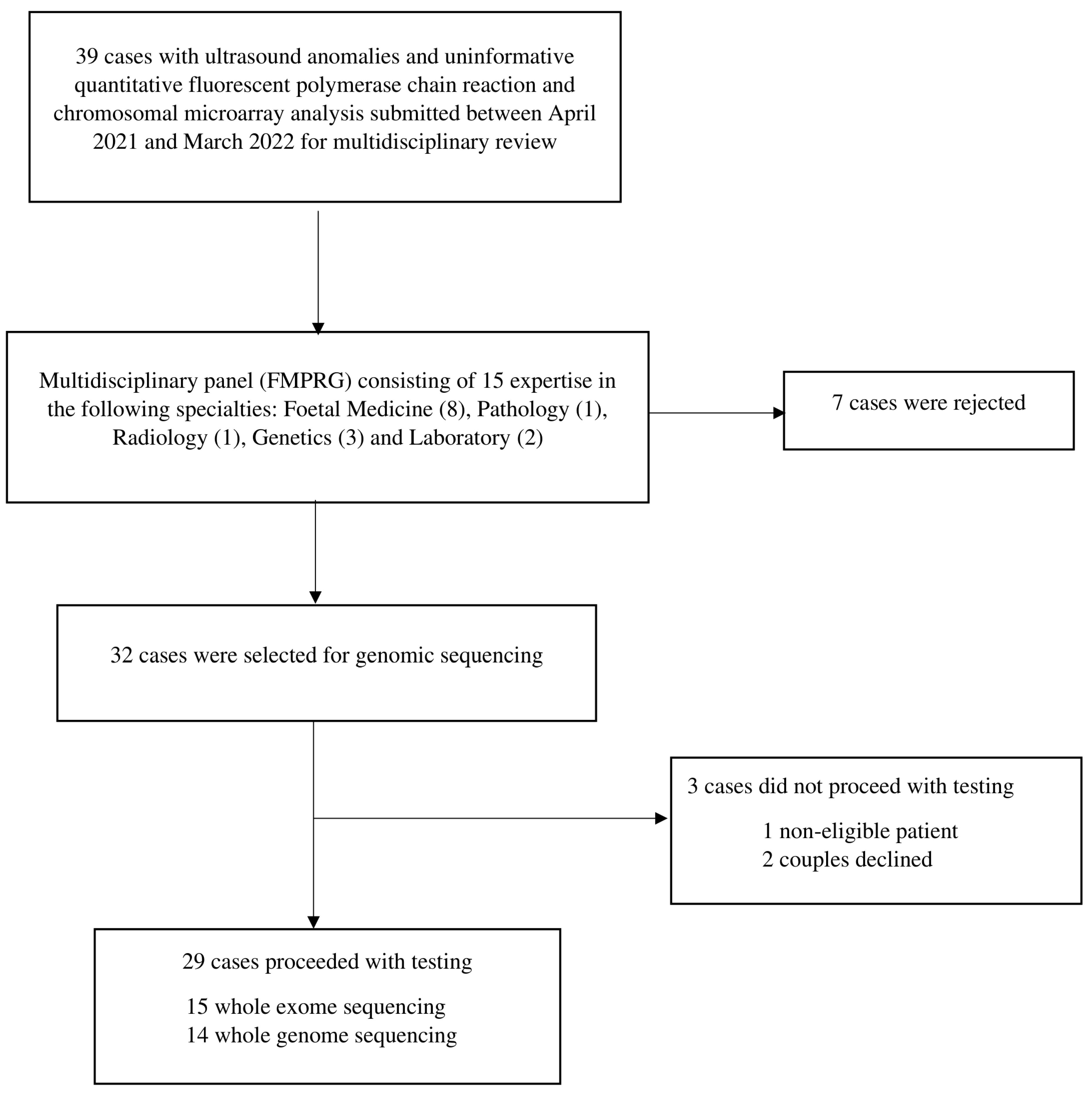
2.1. Study Population and Selection
2.2. WES Procedure and Variant Interpretation
2.3. WGS Procedure and Variant Interpretation
2.4. Post-Test Follow-Up
2.5. Study Data Collection and Analysis
3. Results
3.1. Cohort Characteristics
3.2. Overview of Diagnostic Yield and Prenatal Ultrasound Identified Abnormalities
3.3. Overview of Pregnancy Outcome and Clinical Impact (Table 2 and Table 3)
3.3.1. Pregnancy Outcome
3.3.2. Clinical Impact- Helped Reproductive Decision-Making
3.3.3. Clinical Impact- Guided Perinatal Management
3.3.4. Clinical Impact: Aided Future Family Planning

{kind=link}
| Case | Gestational Age at Test Order | Prenatal Phenotype | Gene | Fetal Sequencing Result | ACMG Classification | Molecular Diagnosis | Inheritance | Novel Variant | Clinical Impact | Pregnancy Outcome | Post-Mortem Result/Postnatal Phenotype | Incidental Finding | Secondary Finding |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ES1 | Post-TOP | USG: absent bilateral lens, microphthalmia, left diaphragmatic hernia, overlapping fingers over bilateral hands | RARB | NM_000965.3:c.1210C>T p.(Gln404Ter), het, de novo | Likely pathogenic | Syndromic microphthalmia-12 | Autosomal dominant | No | Information provision | TOP | Fused eyelids, absent lens of the left eye, left diaphragmatic hernia with herniation of the left lobe of the liver, the stomach, part of the small intestine and spleen into the left hemithorax | NR | NR |
| ES2 | Post-TOP | USG: holoprosencephaly | PPP1R12A | NM_002480.2:c.609delp.(His204Thrfs*12), het, de novo | Pathogenic | Genitourinary and/or brain malformation syndrome | Autosomal dominant | No | Information provision | TOP | Gross examination showed no gross abnormality; declined autopsy | NR | NR |
| ES3 | Post-TOP | USG: micrognathia, overriding aorta, ventricular septal defect, pulmonary stenosis | NIPBL | NM_015384.4:c.2642dup p.(Ser882Ilefs*10), het, de novo | Pathogenic | Cornelia de Lange syndrome 1 | Autosomal dominant | No | Information provision | TOP | Mandibular hypoplasia, cleft palate, ventricular septal defect, overriding aorta, mild pulmonary stenosis, and Meckel’s diverticulum noted in the ileum | NR | NR |
| ES4 | Post-TOP | USG: cystic hygroma, micrognathia, club feet, hydrops fetalis | NEB | NM_001164508.2:c.8712G>A p.(Trp2904Ter), het, mat NM_001164508.2:c.133_146delTCAGAAACTTCCAA p.(Ser45ThrfsTer48), het, pat | Likely pathogenic Likely pathogenic | Arthrogryposis multiplex congenita 6 Nemaline myopathy 2 | Autosomal recessive | Yes No | Familial diagnosis, family planning and counseling | TOP | Gross examination found low-set ears, micrognathia, and club feet with abnormal posture; declined an autopsy | NR | NR |
| ES5 | Post-TOP | USG: microcephaly, sloping forehead, flat occiput MRI: microcephaly, hypoplasia of bilateral frontal lobes and frontal horns, abnormal sulcation pattern with underdeveloped Sylvian fissure, suspected bilateral thalamic fusion and partial agenesis/dysgenesis of the corpus callosum | ASPM | NM_018136.4:c.349C>T, p.(Arg117*), homo, matpat | Pathogenic | Primary microcephaly 5 | Autosomal recessive | No | Familial diagnosis, family planning and counseling | TOP | Gross examination found microcephaly, sloping forehead and flat occiput; declined fetal autopsy | NR | NR |
| ES6 | 16+1 weeks | USG: invisible cerebellum with dilated cisterna magna, cystic hygroma, upper and lower limbs in flexion posture, rocker bottom feet, hydrops fetalis | HRAS | NM_005343.2:c.182G>A p.(Gln61Arg), het, de novo | Pathogenic | Costello syndrome | Autosomal dominant | No | Information provision | Miscarriage at 18 weeks | Limited internal assessment because of extensive autolysis | NR | NR |
| ES7 | 17+4 weeks | USG: cystic hygroma, hydrops fetalis | LZTR1 | NM_006767.3:c.848G>A, p.(Arg283Gln), het, mat | Variant of uncertain significance | Noonan syndrome | Autosomal dominant | No | Reproductive decision, familial diagnosis, family planning and counseling, and neonatal management and follow-up | Livebirth | Delivery at 38 weeks, at 2 months old, with clinical features of Noonan syndrome | NR | NR |
| ES8 | Post-TOP | USG: occipital encephalocele, hypoplastic left heart, bilateral enlarged cystic kidneys, upper limbs and lower limbs short and bowed, feet with polydactyl | - | - | Negative | - | - | - | - | TOP | Encephalocele, hypoplastic thymus, hypoplastic of left heart, polycystic change of bilateral kidneys, feet with polydactyl | NR | NR |
| ES9 | Post-TOP | USG: dysplastic mitral valve, atrioventricular septal defect, smallish distal aorta, persistent left superior vena cava, absent right kidney, ureterocele, single umbilical artery, persistent right umbilical vein | - | - | Negative | - | - | - | - | TOP | Gross examination found a 3 mm skin tag around the anus, no anal opening and pre-axial polydactyly (extra thumb) over left hand; declined fetal autopsy | NR | NR |
| ES10 | Post-TOP | USG: small cerebellum, absent cavum septum pellucidum, small corpus callosum, mild hypertelorism | - | - | Negative | - | - | - | - | TOP | Mild hypertelorism, low-set ears and the marked autolytic change limited the assessment of internal structure of the brain | NR | NR |
| ES11 | Post-TOP | USG: Heterotaxy syndrome with dextrocardia and central liver, right atrial isomerism, unbalanced atrioventricular septal defect, double outlet right ventricle, pulmonary stenosis, infracardiac total anomalous pulmonary venous connection, bilateral superior vena cava | - | - | Negative | - | - | - | - | TOP | Severe hypertelorism with prominent supra-orbital and infraorbital ridges, short nose, carp-sharped mouth, heterotaxy syndrome with dextrocardia and central liver, complex congenital heart disease with hypoplastic left ventricle, atrioventricular septal defect, bilateral superior vena cava, dominant right ventricle with small pulmonary artery, both lungs with 3 lobes and accentuation of other fissures | NR | NR |
| ES12 | Post-IUD | USG: early onset intrauterine growth restriction, oligohydramnios, placentomegaly, single umbilical artery | - | - | Negative | - | - | - | - | Intrauterine death at 27+4 weeks | Weight 0.59 kg, low-set ears and receding jaw | NR | NR |
| ES13 | Post-TOP | USG: bilateral hydrocephalus, small and abnormal cerebellum and deficient vermis, severe micrognathia, bilateral fixed equinovarus with pes cavus, left lower lobe congenital pulmonary airway malformations | - | - | Negative | - | - | - | - | TOP | Declined fetal autopsy | NR | NR |
| ES14 | Post-TOP | USG: hydrops fetalis | - | - | Negative | - | - | - | - | TOP | Hydrops fetalis | NR | NR |
| ES15 | Post-TOP | USG: alobar holoprosencephaly, depressed facial profile, no obvious nose seen, no proboscis, single orbit, hypoplastic left heart, left hand abnormal with second finger shorter and abnormal posture | - | - | Negative | - | - | - | - | TOP | Gross examination found a flat facial profile, absent nose, and shortened second finger of the left hand; declined fetal autopsy | NR | NR |
| Case | Gestational Age at Test Order | Prenatal Phenotype | Gene | Fetal Sequencing Result | ACMG Classification | Molecular Diagnosis | Inheritance | Novel Variant | Clinical Impact | Pregnancy Outcome | Post-Mortem Result/Postnatal Phenotype | Incidental Finding | Secondary Finding |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GS1 | 32+1 weeks | USG: hydrocephalus, periventricular nodular heterotopia, mega cisterna magna, partial dysgenesis of the corpus callosum MRI: diffuse bilateral periventricular grey matter heterotropia, mega cisterna magna, normal corpus callosum | FLNA | NM_001456.3 exon39 c.6368_6369del p.S2123fs, hemi, mat | Likely pathogenic | Periventricular nodular heterotopia | X-linked | No | Familial diagnosis, family planning and counselling, neonatal management and follow-up | Livebirth | Delivery at 37+1 for IUGR, postdelivery MRI showed mild hydrocephalus, periventricular nodular heterotopia, prominent extra-axial cerebrospinal fluid spaces, mega cisterna magna, thinned corpus callosum; at 8 months old, normal growth and development | - | Fetus and father: heterozygous deletion on 16p13.3, including HBA1 and HBA2 (carrier of α thalassemia-1) |
| GS2 | Post-TOP | USG: hypomineralized cranial bone above the base of the skull, short and angulated right femur | COL1A1 | NM_000088.3 exon33 c.2299G>A p.G767S, het, de novo | Pathogenic | Osteogenesis imperfecta | Autosomal dominant | No | Information provision, familial diagnosis, family planning and counselling | TOP | Declined fetal autopsy | - | Father: heterozygous pathogenic variant, c.1747C>T(p.H583Y) in the LDLR gene (autosomal dominant familial hypercholesterolemia); carrier of pathogenic variant G6PC c.248G>A (autosomal recessive glycogen storage disease Ia) |
| GS3 | Post-TOP | USG: agenesis of the corpus callosum, small cerebellum MRI: agenesis of the corpus callosum, small cerebellum | SMARCB1 LAMC3 | NM_003073.5 exon9 c.1130G>A p.R377H, het, de novo NM_006059.4 exon12 c.1963C>T p.R655W, het, mat | Pathogenic Variant of uncertain significance | Coffin-Siris syndrome Malformations of cortical development | Autosomal dominant Autosomal recessive | No No | Information provision, familial diagnosis, family planning and counseling | TOP | Gross examination found bilateral club foot with short big toes, bilateral clenched hands, low-set ears; declined fetal autopsy Placental pathologic finding of single umbilical artery | LAMC3 NM_006059.4 exon18 c.3140G>A p.W1047X, het, pat (likely pathogenic, autosomal recessive malformations of cortical development) | - |
| GS4 | Post-TOP | USG: semilobar holoprosencephaly, bilateral microphthalmia, left lens absent, hypotelorism, bilateral cleft lip and cleft palate, low thoracic scoliosis, suspected hemivertebra, bilateral club foot | RERE GLI2 | NM_012102.4 exon19 c.3385C>G p.Q1129E, het, de novo NM_005270.5 intron13 c.2293+3G>A, het, pat | Likely pathogenic Variant of uncertain significance | Neurodevelopmental disorder with or without anomalies of the brain, eye, or heart Holoprosencephaly 9 | Autosomal dominant Autosomal dominant | No | Information provision, familial diagnosis, family planning and counseling | TOP | Gross examination found bilateral microphthalmia, hypotelorism, bilateral cleft lips and cleft palate, micrognathia, ambiguous genitalia, and left clubfoot; declined fetal autopsy | - | - |
| GS5 | Post-TOP | USG: Tetralogy of Fallot, persistent left superior vena cava, atrioventricular septal defect, horseshoe kidneys, single umbilical artery | KMT2D | NM_003482.4 exon11/55 c.1899dup p.Pro634AlafsTer7, het, de novo | Pathogenic | Kabuki syndrome | Autosomal dominant | Yes | Information provision | TOP | Tetralogy of Fallot with atrioventricular septal defect, horseshoe kidneys with fusion at the lower pole, single umbilical artery | - | - |
| GS6 | 29 weeks | USG: bilateral Sylvian fissure angle severely delayed, asymmetrical diagonal echogenic lines outside the ventricles, suggestive of neuronal migration disorder, aortic stenosis, aortopulmonary window, early onset symmetrical IUGR, oligohydramnios MRI: no structural brain abnormalities | MAP2K2 | NM_030662.3 exon1 c.3G>A p.M1I, het, mat | Variant of uncertain significance | Cranio-facio-cutaneous syndrome | Autosomal dominant | No | Familial diagnosis, family planning and counselling, neonatal management and follow-up | Livebirth | Delivery at 35+2 weeks for abnormal Doppler and IUGR, patent ductus arteriosus with ligation for uncontrolled heart failure on day 20, bovine aortic arch, moderate aortic stenosis, secundum atrial septal defect, right renal stone; at 13 months old, gross motor delay on physiotherapy training, closed atrial septal defect, resolving nephrocalcinosis | - | - |
| GS7 | Post-TOP | USG: hydrops fetalis | COL1A1 COL10A1 | NM_000088.3 exon47 c.3469G>A p.G1157S, het, pat COL10A1 NM_000493.4 exon3c.688C>T p.Q230X, het, pat | Variant of uncertain significance Variant of uncertain significance | Osteogenesis imperfecta Metaphyseal chondrodysplasia, Schmid type | Autosomal dominant Autosomal dominant | No No | Familial diagnosis, family planning and counselling | TOP | Gross examination found hydropic abortus; declined fetal autopsy | KPTN NM_007059.4 intron4 c.450-2A > G, hom, matpat (likely pathogenic, intellectual developmental disorder, autosomal recessive 41) TCF12 NM_207036.2 exon11 c.956C>G p.S319X, het, mat (likely pathogenic, autosomal dominant, craniosynostosis 3) 172.8 Mb of the absence of heterozygosity region | - |
| GS8 | 21+4 weeks | USG: micrognathia | - | - | Negative | - | - | - | Reproductive decision | Livebirth | Delivery at 39+5 weeks, normal newborn examination | - | Fetus and father: heterozygous deletion on 16p13.3, including HBA1 and HBA2 (carrier of α-thalassemia-1) |
| GS9 | 20+3 weeks | USG: megacystis, bilateral dilated renal pelvises | - | - | Negative | - | - | Reproductive decision | Livebirth # | Delivery at 39+3 weeks, at 5 months old, bilateral grade 4 vesicoureteral reflux, posterior urethral valve with bilateral hydronephrosis | - | - | |
| GS10 | 19+2 weeks | USG: megacystis, bilateral dilated renal pelvises, umbilical cord cyst oligohydramnios | - | - | Negative | - | - | Reproductive decision | TOP | Termination of pregnancy in the private sector, postmortem report not available | - | - | |
| GS11 | Post- selective TOP 20+2 weeks | USG: monochorionic, diamniotic twin, with one hydrops with cystic hygroma | - | - | Negative | - | - | Reproductive decision | Selective feticide; co-twin livebirth | Delivery of remaining twin at 41 weeks, normal newborn examination | - | - | |
| GS12 | Post-TOP | USG: cerebellar hypoplasia, cardiomegaly, right ventricular hypertrophy, pericardial effusion | - | - | Negative | - | - | - | TOP | Cerebellar hypoplasia, increased right cardiac ventricular wall thickness, atrial septal defect, pulmonary hypoplasia | |||
| GS13 | Post-TOP | USG: cerebellar hypoplasia, left ventriculomegaly | - | - | Negative | - | - | - | TOP | Hypertelorism, left ventriculomegaly, inconclusive of cerebellar hypoplasia, right thumb hypoplasia | |||
| GS14 | Post-TOP | USG: bilateral borderline ventriculomegaly, agenesis of the corpus callosum MRI: complete agenesis of the corpus callosum | - | - | Negative | - | - | - | TOP | Agenesis of corpus callosum |
3.4. Personnel Involving in Pre-Test and Post-Test Genetic Counselling
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rydberg, C.; Tunón, K. Detection of fetal abnormalities by second-trimester ultrasound screening in a non-selected population. Acta Obstet. Gynecol. Scand. 2017, 96, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Oza, S.; Hogan, D.; Perin, J.; Rudan, I.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: An updated systematic analysis. Lancet 2015, 385, 430–440. [Google Scholar] [CrossRef]
- Carmichael, S.L. Birth defects epidemiology. Eur. J. Med. Genet. 2014, 57, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Toufaily, M.H.; Westgate, M.N.; Lin, A.E.; Holmes, L.B. Causes of Congenital Malformations. Birth Defects Res. 2018, 110, 87–91. [Google Scholar] [CrossRef]
- Lord, J.; McMullan, D.J.; Eberhardt, R.Y.; Rinck, G.; Hamilton, S.J.; Quinlan-Jones, E.; Prigmore, E.; Keelagher, R.; Best, S.K.; Carey, G.K.; et al. Prenatal Assessment of Genomes and Exomes Consortium. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): A cohort study. Lancet 2019, 393, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Petrovski, S.; Aggarwal, V.; Giordano, J.L.; Stosic, M.; Wou, K.; Bier, L.; Spiegel, E.; Brennan, K.; Stong, N.; Jobanputra, V.; et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: A prospective cohort study. Lancet 2019, 393, 758–767. [Google Scholar] [CrossRef]
- Mellis, R.; Oprych, K.; Scotchman, E.; Hill, M.; Chitty, L.S. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: A systematic review and meta-analysis. Prenat. Diagn. 2022, 42, 662–685. [Google Scholar] [CrossRef]
- Pauta, M.; Martinez-Portilla, R.J.; Borrell, A. Diagnostic yield of exome sequencing in fetuses with multisystem malformations: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2022, 59, 715–722. [Google Scholar] [CrossRef]
- Mastromoro, G.; Guadagnolo, D.; Khaleghi Hashemian, N.; Marchionni, E.; Traversa, A.; Pizzuti, A. Molecular Approaches in Fetal Malformations, Dynamic Anomalies and Soft Markers: Diagnostic Rates and Challenges-Systematic Review of the Literature and Meta-Analysis. Diagnostics 2022, 12, 575. [Google Scholar] [CrossRef]
- Van den Veyver, I.B.; Chandler, N.; Wilkins-Haug, L.E.; Wapner, R.J.; Chitty, L.S.; ISPD Board of Directors. International Society for Prenatal Diagnosis Updated Position Statement on the use of genome-wide sequencing for prenatal diagnosis. Prenat. Diagn. 2022, 42, 796–803. [Google Scholar] [CrossRef]
- Monaghan, K.G.; Leach, N.T.; Pekarek, D.; Prasad, P.; Rose, N.C.; ACMG Professional Practice and Guidelines Committee. The use of fetal exome sequencing in prenatal diagnosis: A points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Greenfeld, E.; Watkins, N.; Belesiotis, P.; Zaidi, S.H.; Marshall, C.; Thiruvahindrapuram, B.; Shannon, P.; Roifman, M.; Chong, K.; et al. Diagnostic yield of genome sequencing for prenatal diagnosis of fetal structural anomalies. Prenat. Diagn. 2022, 42, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, Z.; Sun, J.; Liu, L.; Zhou, X.; Liu, F.; Xing, Y.; Cui, S.; Xiong, S.; Liu, X.; et al. Whole Genome Sequencing in the Evaluation of Fetal Structural Anomalies: A Parallel Test with Chromosomal Microarray Plus Whole Exome Sequencing. Genes 2021, 12, 376. [Google Scholar] [CrossRef] [PubMed]
- Choy, K.W.; Wang, H.; Shi, M.; Chen, J.; Yang, Z.; Zhang, R.; Yan, H.; Wang, Y.; Chen, S.; Chau, M.H.K.; et al. Prenatal Diagnosis of Fetuses With Increased Nuchal Translucency by Genome Sequencing Analysis. Front. Genet. 2019, 10, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Yang, Y.; Wen, H.; Wang, B.; Zhang, T.; Li, S. Abnormal Sylvian fissure at 20-30 weeks as indicator of malformations of cortical development: Role of prenatal whole-genome sequencing. Ultrasound Obstet. Gynecol. 2022, 59, 552–555. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Yang, Y.K.; Liang, Y.; Zhang, T.J.; Liang, N.; Yang, L.M.; Li, S.J.; Shan, D.; Wu, Q.Q. Prenatal diagnosis of fetal skeletal dysplasia using targeted next-generation sequencing: An analysis of 30 cases. Diagn. Pathol. 2019, 14, 76. [Google Scholar] [CrossRef] [Green Version]
- Castleman, J.S.; Wall, E.; Allen, S.; Williams, D.; Doyle, S.; Kilby, M.D. The prenatal exome-a door to prenatal diagnostics? Expert Rev. Mol. Diagn. 2021, 21, 465–474. [Google Scholar] [CrossRef]
- Normand, E.A.; Braxton, A.; Nassef, S.; Ward, P.A.; Vetrini, F.; He, W.; Patel, V.; Qu, C.; Westerfield, L.E.; Stover, S.; et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med. 2018, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- de Koning, M.A.; Hoffer, M.; Nibbeling, E.; Bijlsma, E.K.; Toirkens, M.; Adama-Scheltema, P.N.; Verweij, E.J.; Veenhof, M.B.; Santen, G.; Peeters-Scholte, C. Prenatal exome sequencing: A useful tool for the fetal neurologist. Clin. Genet. 2022, 101, 65–77. [Google Scholar] [CrossRef]
- Corsten-Janssen, N.; Bouman, K.; Diphoorn, J.; Scheper, A.J.; Kinds, R.; El Mecky, J.; Breet, H.; Verheij, J.; Suijkerbuijk, R.; Duin, L.K.; et al. A prospective study on rapid exome sequencing as a diagnostic test for multiple congenital anomalies on fetal ultrasound. Prenat. Diagn. 2020, 40, 1300–1309. [Google Scholar] [CrossRef]
- Tolusso, L.K.; Hazelton, P.; Wong, B.; Swarr, D.T. Beyond diagnostic yield: Prenatal exome sequencing results in maternal, neonatal, and familial clinical management changes. Genet. Med. 2021, 23, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Deden, C.; Neveling, K.; Zafeiropopoulou, D.; Gilissen, C.; Pfundt, R.; Rinne, T.; de Leeuw, N.; Faas, B.; Gardeitchik, T.; Sallevelt, S.; et al. Rapid whole exome sequencing in pregnancies to identify the underlying genetic cause in fetuses with congenital anomalies detected by ultrasound imaging. Prenat. Diagn. 2020, 40, 972–983. [Google Scholar] [CrossRef] [PubMed]
- de Koning, M.A.; Haak, M.C.; Adama van Scheltema, P.N.; Peeters-Scholte, C.; Koopmann, T.T.; Nibbeling, E.; Aten, E.; den Hollander, N.S.; Ruivenkamp, C.; Hoffer, M.; et al. From diagnostic yield to clinical impact: A pilot study on the implementation of prenatal exome sequencing in routine care. Genet. Med. 2019, 21, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, E.; Haworth, A.; Ive, L.; Dubis, R.; Savage, H.; Serra, E.; Kenny, J.; Elmslie, F.; Greco, E.; Thilaganathan, B.; et al. A report on the impact of rapid prenatal exome sequencing on the clinical management of 52 ongoing pregnancies: A retrospective review. BJOG 2021, 128, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- NHS. (n.d.). NHS Choices. Retrieved 20 October 2022. Available online: https://www.england.nhs.uk/publication/national-genomic-testdirectories/25.https://www.england.nhs.uk/publication/national-genomic-test-directories/ (accessed on 25 October 2022).
- Sparks, T.N.; Lianoglou, B.R.; Adami, R.R.; Pluym, I.D.; Holliman, K.; Duffy, J.; Downum, S.L.; Patel, S.; Faubel, A.; Boe, N.M.; et al. Exome Sequencing for Prenatal Diagnosis in Nonimmune Hydrops Fetalis. N. Engl. J. Med. 2020, 383, 1746–1756. [Google Scholar] [CrossRef]
- Al-Kouatly, H.B.; Makhamreh, M.M.; Rice, S.M.; Smith, K.; Harman, C.; Quinn, A.; Valcarcel, B.N.; Firman, B.; Liu, R.; Hegde, M.; et al. High diagnosis rate for nonimmune hydrops fetalis with prenatal clinical exome from the Hydrops-Yielding Diagnostic Results of Prenatal Sequencing (HYDROPS) Study. Genet. Med. 2021, 23, 1325–1333. [Google Scholar] [CrossRef]
- Mone, F.; Eberhardt, R.Y.; Hurles, M.E.; Mcmullan, D.J.; Maher, E.R.; Lord, J.; Chitty, L.S.; Dempsey, E.; Homfray, T.; Giordano, J.L.; et al. Fetal hydrops and the Incremental yield of Next-generation sequencing over standard prenatal Diagnostic testing (FIND) study: Prospective cohort study and meta-analysis. Ultrasound Obstet. Gynecol. 2021, 58, 509–518. [Google Scholar] [CrossRef]
- Kelley, J.; McGillivray, G.; Meagher, S.; Hui, L. Increased nuchal translucency after low-risk noninvasive prenatal testing: What should we tell prospective parents? Prenat. Diagn. 2021, 41, 1305–1315. [Google Scholar] [CrossRef]
- Chen, M.H.; Walsh, C.A. FLNA Deficiency. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Eds.; 8 October 2002 [Updated 30 September 2021]; University of Washington: Seattle, WA, USA, 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1213/ (accessed on 25 October 2022).
- Best, S.; Wou, K.; Vora, N.; Van der Veyver, I.B.; Wapner, R.; Chitty, L.S. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn. 2018, 38, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Westerfield, L.; Darilek, S.; van den Veyver, I.B. Counseling Challenges with Variants of Uncertain Significance and Incidental Findings in Prenatal Genetic Screening and Diagnosis. J. Clin. Med. 2014, 3, 1018–1032. [Google Scholar] [CrossRef]
- Pratt, M.; Garritty, C.; Thuku, M.; Esmaeilisaraji, L.; Hamel, C.; Hartley, T.; Millar, K.; Skidmore, B.; Dougan, S.; Armour, C.M. Application of exome sequencing for prenatal diagnosis: A rapid scoping review. Genet. Med. 2020, 22, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Vora, N.L.; Gilmore, K.; Brandt, A.; Gustafson, C.; Strande, N.; Ramkissoon, L.; Hardisty, E.; Foreman, A.; Wilhelmsen, K.; Owen, P.; et al. Correction: An approach to integrating exome sequencing for fetal structural anomalies into clinical practice. Genet. Med. 2020, 22, 1426. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, B.B.; Klein, W.; Lewis, K.L.; Fisher, T.C.; Wright, M.F.; Biesecker, L.G.; Han, P.K. How do research participants perceive “uncertainty” in genome sequencing? Genet. Med. 2014, 16, 977–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amor, D.J.; Chitty, L.S.; Van den Veyver, I.B. Current controversies in prenatal diagnosis 2: The 59 genes ACMG recommends reporting as secondary findings when sequencing postnatally should be reported when detected on fetal (and parental) sequencing. Prenat. Diagn. 2020, 40, 1508–1514. [Google Scholar] [CrossRef]
- Marokakis, S.; Kasparian, N.A.; Kennedy, S.E. Prenatal counselling for congenital anomalies: A systematic review. Prenat. Diagn. 2016, 36, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Skari, H.; Malt, U.F.; Bjornland, K.; Egeland, T.; Haugen, G.; Skreden, M.; Dalholt Björk, M.; Bjornstad Ostensen, A.; Emblem, R. Prenatal diagnosis of congenital malformations and parental psychological distress--a prospective longitudinal cohort study. Prenat. Diagn. 2006, 26, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Talati, A.N.; Gilmore, K.L.; Hardisty, E.E.; Lyerly, A.D.; Rini, C.; Vora, N.L. Impact of prenatal exome sequencing for fetal genetic diagnosis on maternal psychological outcomes and decisional conflict in a prospective cohort. Genet. Med. 2021, 23, 713–719. [Google Scholar] [CrossRef]

| Characteristic | Value * |
|---|---|
| Ethnicity | |
| Chinese | 26 (89.7) |
| South Asian | 3 (10.3) |
| Maternal age (year) | 33 (25–39) |
| Educational level | |
| Primary | 0 (0) |
| Secondary | 9 (31.0) |
| Tertiary | 18 (62.1) |
| Unknown | 2 (6.9) |
| Married | 28 (96.6) |
| Consanguinity | 2 (6.9) |
| Family history of genetic problems | 1 (3.4) |
| Previous pregnancy/child of genetic problems | 2 (6.9) |
| Nulliparity | 19 (65.5) |
| History of pregnancy loss | 6 (20.7) |
| History of pregnancy termination | 4 (13.8) |
| Planned pregnancy | 23 (79.3) |
| Conception | |
| Natural | 26 (89.7) |
| Assisted | 3 (10.3) |
| Number of fetuses | |
| Singleton | 28 (96.6) |
| Twin | 1 (3.4) |
| Type of testing | |
| Whole exome sequencing (WES) | 15 (51.7) |
| Whole genome sequencing (WGS) | 14 (48.3) |
| Trio analysis | 29 (100) |
| Invasive diagnostic procedure | |
| No | 3 (10.3) |
| Chorionic villus sampling | 7 (24.1) |
| Amniocentesis | 19 (65.5) |
| Source of fetal DNA | |
| Chorionic villi | 6 (20.7) |
| Amniocytes | 19 (65.5) |
| Products of conception | 3 (10.3) |
| Fetal blood/tissue | 1 (3.4) |
| Prenatal phenotype at test request | |
| Single system | 11 (37.9) |
| - Brain malformation | 7 (24.1) |
| - Renal malformation | 2 (6.9) |
| - Skeletal malformation | 1 (3.4) |
| - Facial malformation | 1 (3.4) |
| Multiple systems | 11 (37.9) |
| Intrauterine growth restriction | 1 (3.4) |
| Hydrops fetalis | 6 (20.7) |
| Timing of test request | |
| Before 24 weeks of pregnancy | 6 (20.7) |
| After 24 weeks of pregnancy | 2 (6.9) |
| After intrauterine death | 1 (3.4) |
| After the termination of pregnancy | 20 (69.0) |
| Report turnaround time (day) during pregnancy | 19.5 (13–31) |
| WES results (genetic variant) | |
| Pathogenic/Likely pathogenic | 7 (46.7) |
| Variant of uncertain significance | 1 (6.7) |
| Negative | 8 (53.3) |
| WGS results (genetic variant) | |
| Pathogenic/Likely pathogenic | 5 (35.7) |
| Variant of uncertain significance | 5 (35.7) |
| Negative | 7 (50.0) |
| Incidental finding | 3 (21.4) |
| Secondary finding | 4 (28.6) |
| Pregnancy outcome | |
| Livebirth | 5 (17.2) |
| Intrauterine death | 1 (3.4) |
| Miscarriage | 1 (3.4) |
| Termination of pregnancy | 22 (75.9) |
| Neonatal death | 0 (0) |
| Personnel conducting pre-test counseling | |
| Midwife | 0 (0) |
| Maternal-fetal medicine specialist/obstetrician | 27 (93.1) |
| Clinical geneticist | 2 (6.9) |
| Personnel conducting post-test counseling | |
| Midwife | 1 (3.4) |
| Maternal-fetal medicine specialist/obstetrician | 17 (58.6) |
| Clinical geneticist | 10 (34.5) |
| Not available | 1 (3.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
So, P.L.; Hui, A.S.Y.; Ma, T.W.L.; Shu, W.; Hui, A.P.W.; Kong, C.W.; Lo, T.K.; Kan, A.N.C.; Kan, E.Y.L.; Chong, S.C.; et al. Implementation of Public Funded Genome Sequencing in Evaluation of Fetal Structural Anomalies. Genes 2022, 13, 2088. https://doi.org/10.3390/genes13112088
So PL, Hui ASY, Ma TWL, Shu W, Hui APW, Kong CW, Lo TK, Kan ANC, Kan EYL, Chong SC, et al. Implementation of Public Funded Genome Sequencing in Evaluation of Fetal Structural Anomalies. Genes. 2022; 13(11):2088. https://doi.org/10.3390/genes13112088
Chicago/Turabian StyleSo, Po Lam, Annie Shuk Yi Hui, Teresa Wei Ling Ma, Wendy Shu, Amelia Pui Wah Hui, Choi Wah Kong, Tsz Kin Lo, Amanda Nim Chi Kan, Elaine Yee Ling Kan, Shuk Ching Chong, and et al. 2022. "Implementation of Public Funded Genome Sequencing in Evaluation of Fetal Structural Anomalies" Genes 13, no. 11: 2088. https://doi.org/10.3390/genes13112088
APA StyleSo, P. L., Hui, A. S. Y., Ma, T. W. L., Shu, W., Hui, A. P. W., Kong, C. W., Lo, T. K., Kan, A. N. C., Kan, E. Y. L., Chong, S. C., Chung, B. H. Y., Luk, H. M., Choy, K. W., Kan, A. S. Y., & Leung, W. C. (2022). Implementation of Public Funded Genome Sequencing in Evaluation of Fetal Structural Anomalies. Genes, 13(11), 2088. https://doi.org/10.3390/genes13112088