

Identification and Validation of UPF1 as a Novel Prognostic Biomarker in Renal Clear Cell Carcinoma

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Single-Cell RNA Sequencing Data Preprocessing
2.3. UPF1 Expression Analysis and Survival Analysis
2.4. Immunological Features of the TME in ccRCC
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.6. Analysis of UPF1 in Pan-Cancer
2.7. Patients and Samples
2.8. qRT-PCR and IHC
2.9. Cell Culture and Transfection
2.10. Western Blotting
2.11. Cell Counting Kit-8 Assay
2.12. Migration and Invasion Assays
2.13. Statistics
3. Results
3.1. UPF1 Has Low Expression in ccRCCs and Was Correlated with Poor Prognosis
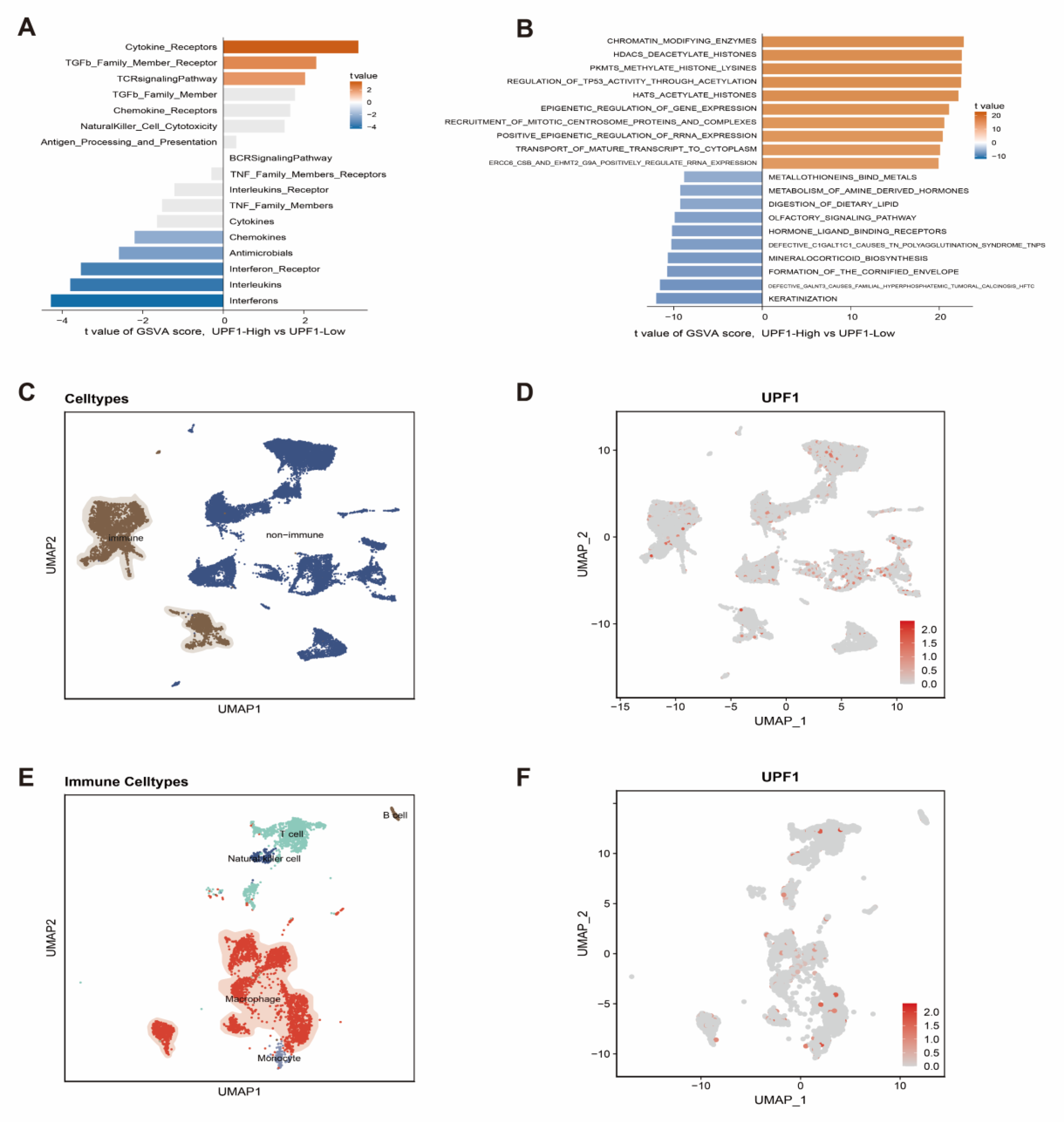
3.2. The Effects of UPF1 in the ccRCC Tumor Microenvironment
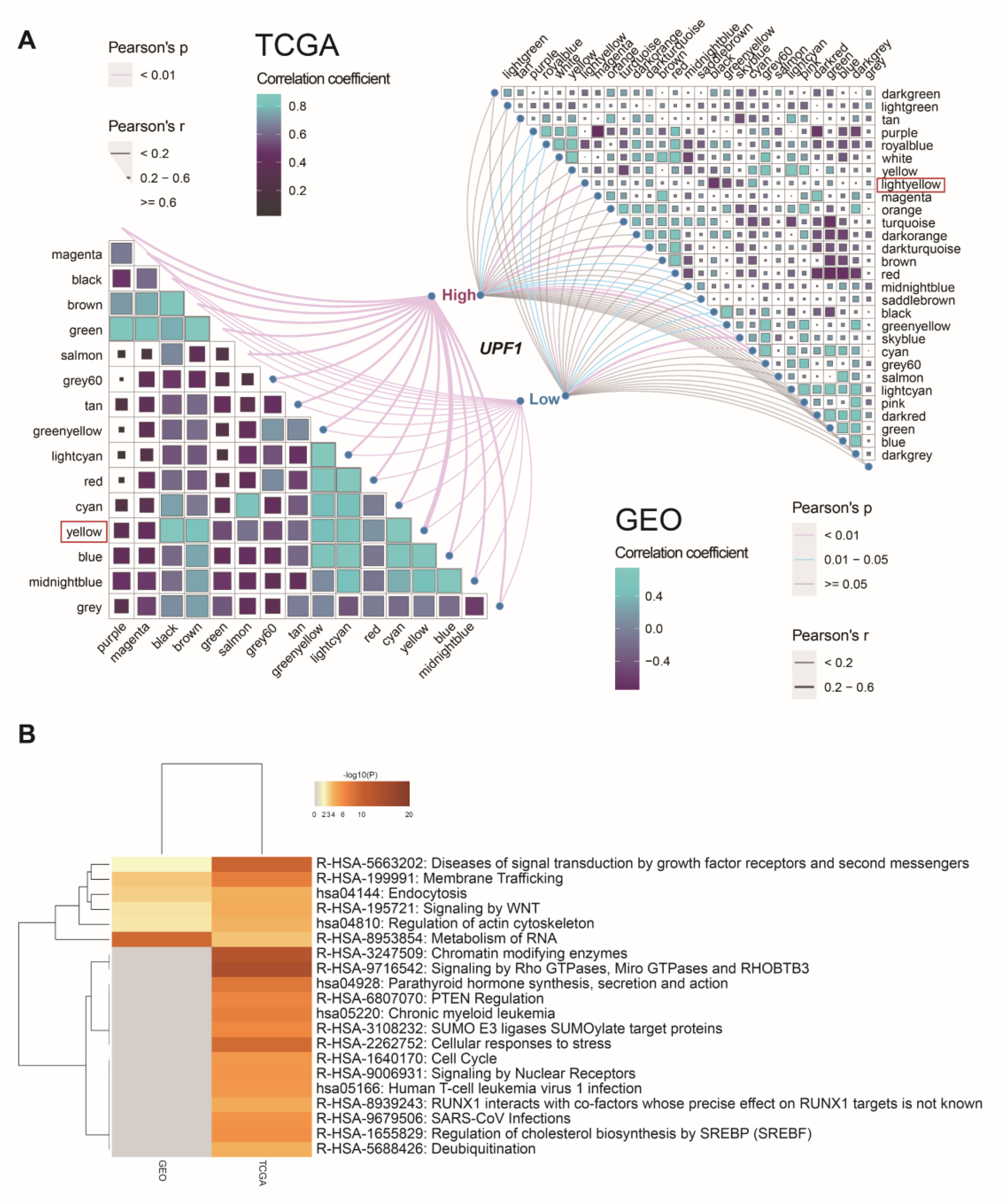
3.3. Screen Key Modules and Co-Expression Genes of UPF1
3.4. Experiments to Validate the Expression and Functions of UPF1
3.5. Analysis of UPF1 in Pan-Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, H.T.; McGovern, F.J. Renal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2477–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, N.; Drake, C.G. Kidney Cancer: An Overview of Current Therapeutic Approaches. Urol. Clin. North Am. 2020, 47, 419–431. [Google Scholar] [CrossRef]
- Yamashita, A.; Ohnishi, T.; Kashima, I.; Taya, Y.; Ohno, S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 2001, 15, 2215–2228. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Karam, R.; Zhou, Y.; Su, F.; Ji, Y.; Li, G.; Xu, G.; Lu, L.; Wang, C.; Song, M.; et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat. Med. 2014, 20, 596–598. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Hwang, Y.; Kim, T.H.; Jeong, J.; Choi, D.; Hwang, J. UPF1 Inhibits Hepatocellular Carcinoma Growth through DUSP1/p53 Signal Pathway. Biomedicines 2022, 10, 793. [Google Scholar] [CrossRef]
- Li, L.; Geng, Y.; Feng, R.; Zhu, Q.; Miao, B.; Cao, J.; Fei, S. The Human RNA Surveillance Factor UPF1 Modulates Gastric Cancer Progression by Targeting Long Non-Coding RNA MALAT1. Cell. Physiol. Biochem. 2017, 42, 2194–2206. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Plank, T.D.; Su, F.; Shi, X.; Liu, C.; Ji, Y.; Li, S.; Huynh, A.; Shi, C.; Zhu, B.; et al. The nonsense-mediated RNA decay pathway is disrupted in inflammatory myofibroblastic tumors. J. Clin. Investig. 2016, 126, 3058–3062. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.B.; Wu, Y.J.; Luo, J.N.; Hu, X.N.; Yuan, Z.N.; Li, G.; Wang, Y.W.; Yao, G.D.; Ge, X.F. Knockdown of long noncoding RNA DLX6-AS1 inhibits migration and invasion of thyroid cancer cells by upregulating UPF1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8246. [Google Scholar] [CrossRef]
- Pei, C.L.; Fei, K.L.; Yuan, X.Y.; Gong, X.J. LncRNA DANCR aggravates the progression of ovarian cancer by downregulating UPF1. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10657–10663. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.H.; Wang, Z.Y.; Li, Z.Y. LncRNA PVT1 aggravates the progression of glioma via downregulating UPF1. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8956–8963. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; He, Q.; Liu, Y.; Liu, X.; Zheng, J.; Ma, J.; Liu, L.; Li, H.; Li, Z.; Xue, Y. UPF1 regulates the malignant biological behaviors of glioblastoma cells via enhancing the stability of Linc-00313. Cell Death Dis. 2019, 10, 629. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Cao, D.; Sha, J.; Zhu, X.; Chen, D. LncRNA ZFPM2-AS1 promotes lung adenocarcinoma progression by interacting with UPF1 to destabilize ZFPM2. Mol. Oncol. 2020, 14, 1074–1088. [Google Scholar] [CrossRef] [Green Version]
- von Roemeling, C.A.; Radisky, D.C.; Marlow, L.A.; Cooper, S.J.; Grebe, S.K.; Anastasiadis, P.Z.; Tun, H.W.; Copland, J.A. Neuronal pentraxin 2 supports clear cell renal cell carcinoma by activating the AMPA-selective glutamate receptor-4. Cancer Res. 2014, 74, 4796–4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, M.B.; Le Calvez-Kelm, F.; Abedi-Ardekani, B.; Byrnes, G.; Durand, G.; Carreira, C.; Michelon, J.; Janout, V.; Holcatova, I.; Foretova, L.; et al. Integrative genome-wide gene expression profiling of clear cell renal cell carcinoma in Czech Republic and in the United States. PLoS ONE 2013, 8, e57886. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Narayanan, S.P.; Mannan, R.; Raskind, G.; Wang, X.; Vats, P.; Su, F.; Hosseini, N.; Cao, X.; Kumar-Sinha, C.; et al. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc. Natl. Acad. Sci. USA 2021, 118, e2103240118. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, T.A.; Liu, M.; et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 2021, 39, 662–677.e666. [Google Scholar] [CrossRef]
- Williams, D.W.; Greenwell-Wild, T.; Brenchley, L.; Dutzan, N.; Overmiller, A.; Sawaya, A.P.; Webb, S.; Martin, D.; Hajishengallis, G.; Divaris, K.; et al. Human oral mucosa cell atlas reveals a stromal-neutrophil axis regulating tissue immunity. Cell 2021, 184, 4090–4104.e4015. [Google Scholar] [CrossRef]
- Zhong, W.; Liu, H.; Li, F.; Lin, Y.; Ye, Y.; Xu, L.; Li, S.; Chen, H.; Li, C.; Lin, Y.; et al. Elevated expression of LIF predicts a poor prognosis and promotes cell migration and invasion of clear cell renal cell carcinoma. Front. Oncol. 2022, 12, 934128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racle, J.; Gfeller, D. EPIC: A Tool to Estimate the Proportions of Different Cell Types from Bulk Gene Expression Data. Methods Mol. Biol. 2020, 2120, 233–248. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Andorf, S.; Gomes, L.; Dunn, P.; Schaefer, H.; Pontius, J.; Berger, P.; Desborough, V.; Smith, T.; Campbell, J.; et al. ImmPort: Disseminating data to the public for the future of immunology. Immunol. Res. 2014, 58, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Taminau, J.; Meganck, S.; Lazar, C.; Steenhoff, D.; Coletta, A.; Molter, C.; Duque, R.; Schaetzen, V.D.; Weiss Solís, D.Y.; Bersini, H.; et al. Unlocking the potential of publicly available microarray data using inSilicoDb and inSilicoMerging R/Bioconductor packages. BMC Bioinform. 2012, 13, 335. [Google Scholar] [CrossRef]
- Chen, C.; Grennan, K.; Badner, J.; Zhang, D.; Gershon, E.; Jin, L.; Liu, C. Removing batch effects in analysis of expression microarray data: An evaluation of six batch adjustment methods. PLoS ONE 2011, 6, e17238. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Song, Z.; Zhong, X.; Huang, M.; Shen, D.; Gao, P.; Qian, X.; Wang, M.; He, X.; Wang, T.; et al. Sangerbox: A comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta 2022, 1, e36. [Google Scholar] [CrossRef]
- Zhang, W.T.; Gong, Y.M.; Zhang, C.Y.; Pan, J.S.; Huang, T.; Li, Y.X. A Novel Cuprotosis-Related Gene FDX1 Signature for Overall Survival Prediction in Clear Cell Renal Cell Carcinoma Patients. Biomed Res. Int. 2022, 2022, 9196540. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, H.; Gao, C.; Chen, J.; Li, H.; Meng, Z.; Bai, J.; Shen, Q.; Wu, H.; Yin, T. Hyperglycemia Enhances Immunosuppression and Aerobic Glycolysis of Pancreatic Cancer Through Upregulating Bmi1-UPF1-HK2 Pathway. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1146–1165. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Maquat, L.E. Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: To die or not to die, that is the question. Curr. Opin. Genet. Dev. 2011, 21, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Karam, R.; Wengrod, J.; Gardner, L.B.; Wilkinson, M.F. Regulation of nonsense-mediated mRNA decay: Implications for physiology and disease. Biochim. Biophys. Acta 2013, 1829, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Lin, J.; Liu, J.; Huang, M.; Zhong, Y.; Liang, B.; Song, X.; Gu, S.; Chang, X.; Huang, D.; et al. A novel lncRNA NR4A1AS up-regulates orphan nuclear receptor NR4A1 expression by blocking UPF1-mediated mRNA destabilization in colorectal cancer. Clin. Sci. 2019, 133, 1457–1473. [Google Scholar] [CrossRef]
- Chang, L.; Li, C.; Guo, T.; Wang, H.; Ma, W.; Yuan, Y.; Liu, Q.; Ye, Q.; Liu, Z. The human RNA surveillance factor UPF1 regulates tumorigenesis by targeting Smad7 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; You, Y.; Zhu, Z. The human RNA surveillance factor Up-frameshift 1 inhibits hepatic cancer progression by targeting MRP2/ABCC2. Biomed. Pharmacother. 2017, 92, 365–372. [Google Scholar] [CrossRef]
- Schmiegel, W.; Schmielau, J.; Henne-Bruns, D.; Juhl, H.; Roeder, C.; Buggisch, P.; Onur, A.; Kremer, B.; Kalthoff, H.; Jensen, E.V. Cytokine-mediated enhancement of epidermal growth factor receptor expression provides an immunological approach to the therapy of pancreatic cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 12622–12626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, K.; Kawakami, M.; Puri, R.K. Cytokine receptor as a sensitizer for targeted cancer therapy. Anticancer Drugs 2002, 13, 693–699. [Google Scholar] [CrossRef]
- Scheller, J.; Engelowski, E.; Moll, J.M.; Floss, D.M. Immunoreceptor Engineering and Synthetic Cytokine Signaling for Therapeutics. Trends Immunol. 2019, 40, 258–272. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, K.; Zhang, K.; Duan, S.; Chen, M.; Zhao, N.; Xu, F.J. Orchestrated Yolk-Shell Nanohybrids Regulate Macrophage Polarization and Dendritic Cell Maturation for Oncotherapy with Augmented Antitumor Immunity. Adv. Mater. 2022, 34, e2108263. [Google Scholar] [CrossRef] [PubMed]
- Lawir, D.F.; Sikora, K.; O’Meara, C.P.; Schorpp, M.; Boehm, T. Pervasive changes of mRNA splicing in upf1-deficient zebrafish identify rpl10a as a regulator of T cell development. Proc. Natl. Acad. Sci. USA 2020, 117, 15799–15808. [Google Scholar] [CrossRef] [PubMed]
- Bokhari, A.; Jonchere, V.; Lagrange, A.; Bertrand, R.; Svrcek, M.; Marisa, L.; Buhard, O.; Greene, M.; Demidova, A.; Jia, J.; et al. Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability. Oncogenesis 2018, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usuki, F.; Yamashita, A.; Fujimura, M. Environmental stresses suppress nonsense-mediated mRNA decay (NMD) and affect cells by stabilizing NMD-targeted gene expression. Sci. Rep. 2019, 9, 1279. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Li, H.; Chang, W.; Zhong, L.; Zhang, L.; Wen, Z.; Mai, S. Identification and Validation of UPF1 as a Novel Prognostic Biomarker in Renal Clear Cell Carcinoma. Genes 2022, 13, 2166. https://doi.org/10.3390/genes13112166
Wu C, Li H, Chang W, Zhong L, Zhang L, Wen Z, Mai S. Identification and Validation of UPF1 as a Novel Prognostic Biomarker in Renal Clear Cell Carcinoma. Genes. 2022; 13(11):2166. https://doi.org/10.3390/genes13112166
Chicago/Turabian StyleWu, Chun, Hongmu Li, Wuguang Chang, Leqi Zhong, Lin Zhang, Zhesheng Wen, and Shijuan Mai. 2022. "Identification and Validation of UPF1 as a Novel Prognostic Biomarker in Renal Clear Cell Carcinoma" Genes 13, no. 11: 2166. https://doi.org/10.3390/genes13112166
APA StyleWu, C., Li, H., Chang, W., Zhong, L., Zhang, L., Wen, Z., & Mai, S. (2022). Identification and Validation of UPF1 as a Novel Prognostic Biomarker in Renal Clear Cell Carcinoma. Genes, 13(11), 2166. https://doi.org/10.3390/genes13112166