
Evolutionary Genetic Signatures of Selection on Bone-Related Variation within Human and Chimpanzee Populations

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Population Datasets
2.2. Non-Human Primate Population Datasets
2.3. Statistical Analyses of COL1A1 Amino Acid Variation
2.4. Statistical Analyses of COL1A1 Intronic Variation
3. Results
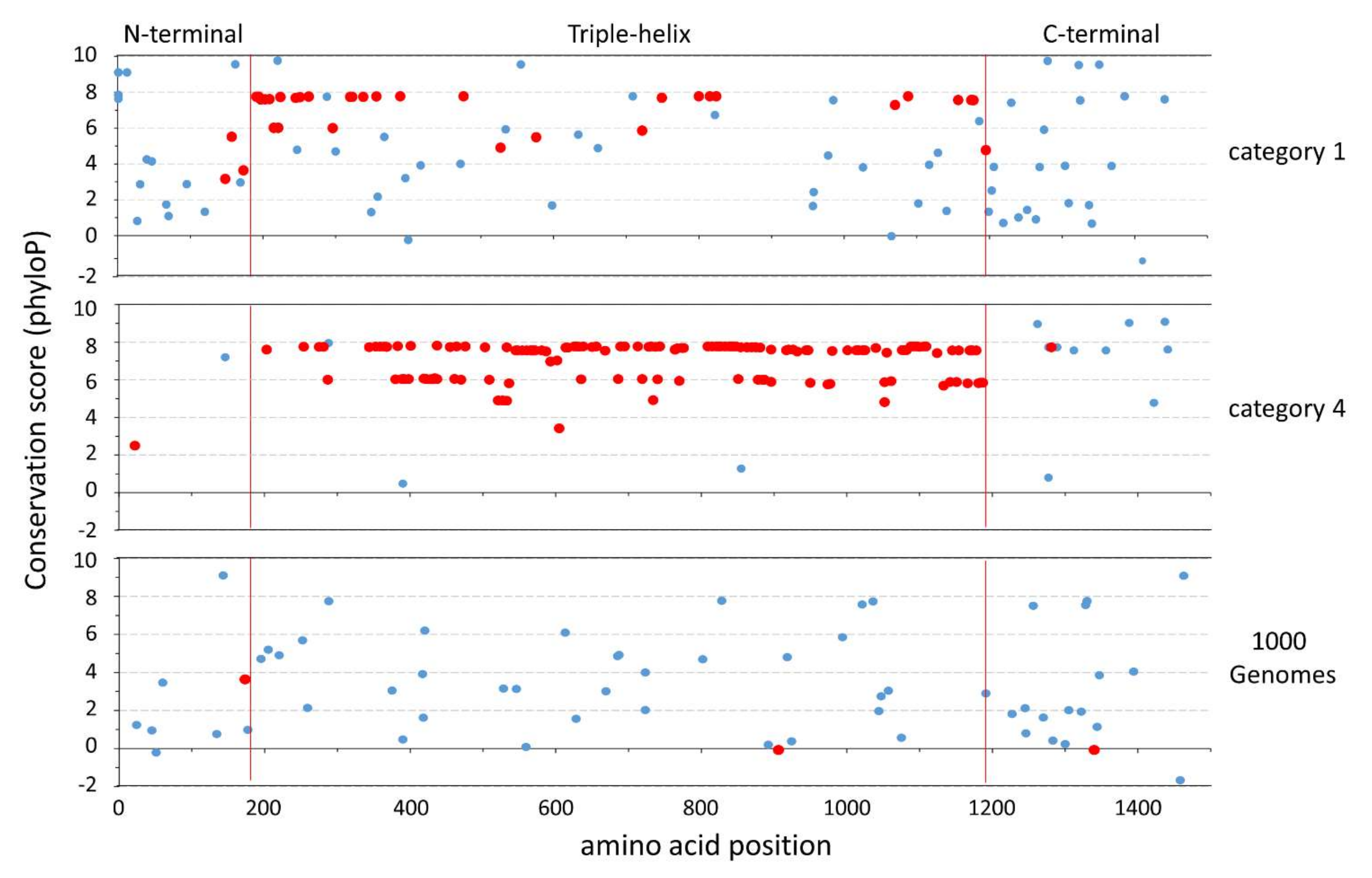
3.1. Contrasts of Amino Acid Variation between the DAM and 1000G Datasets
3.2. Chimpanzee Amino Acid Variation
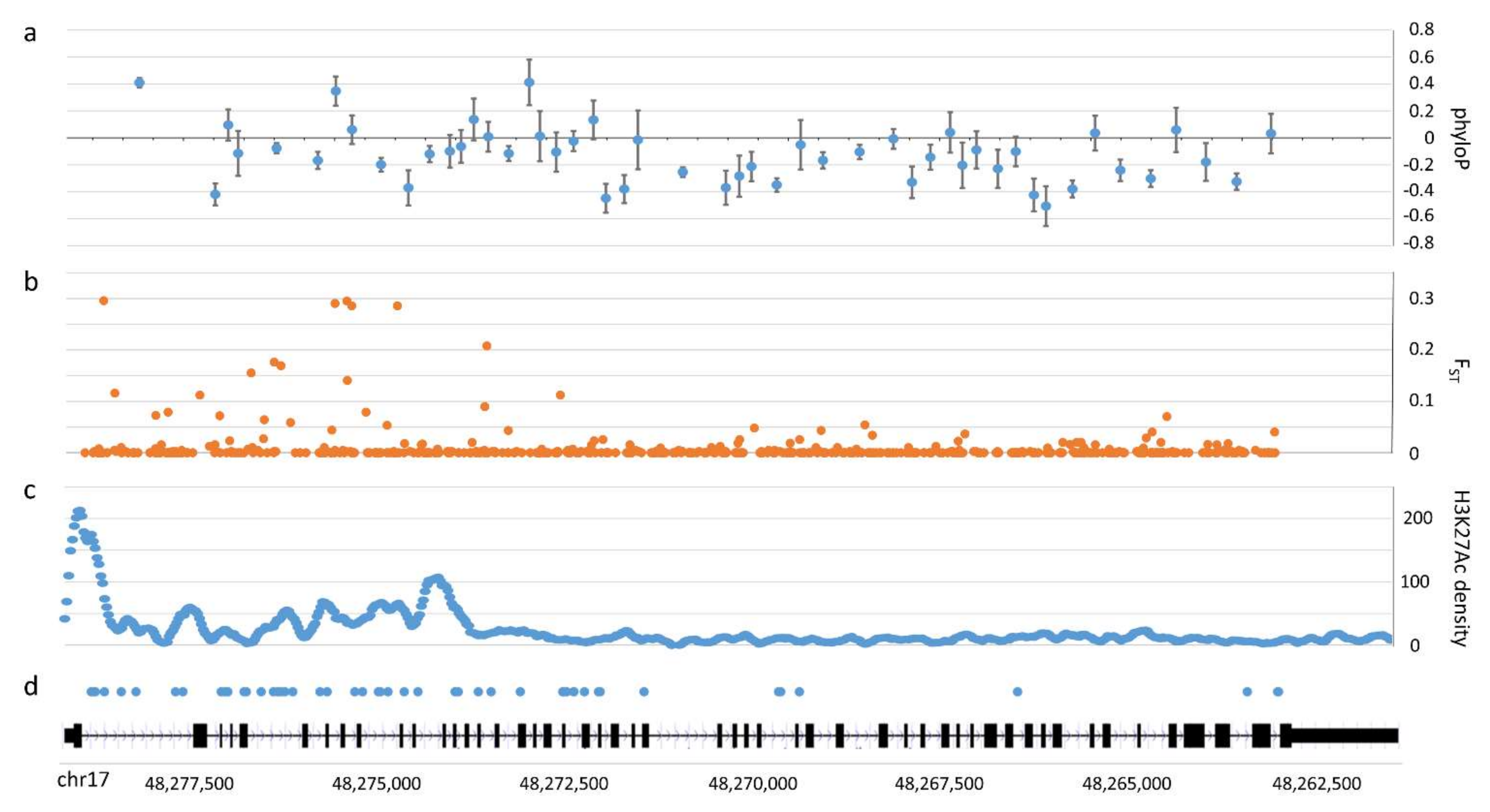
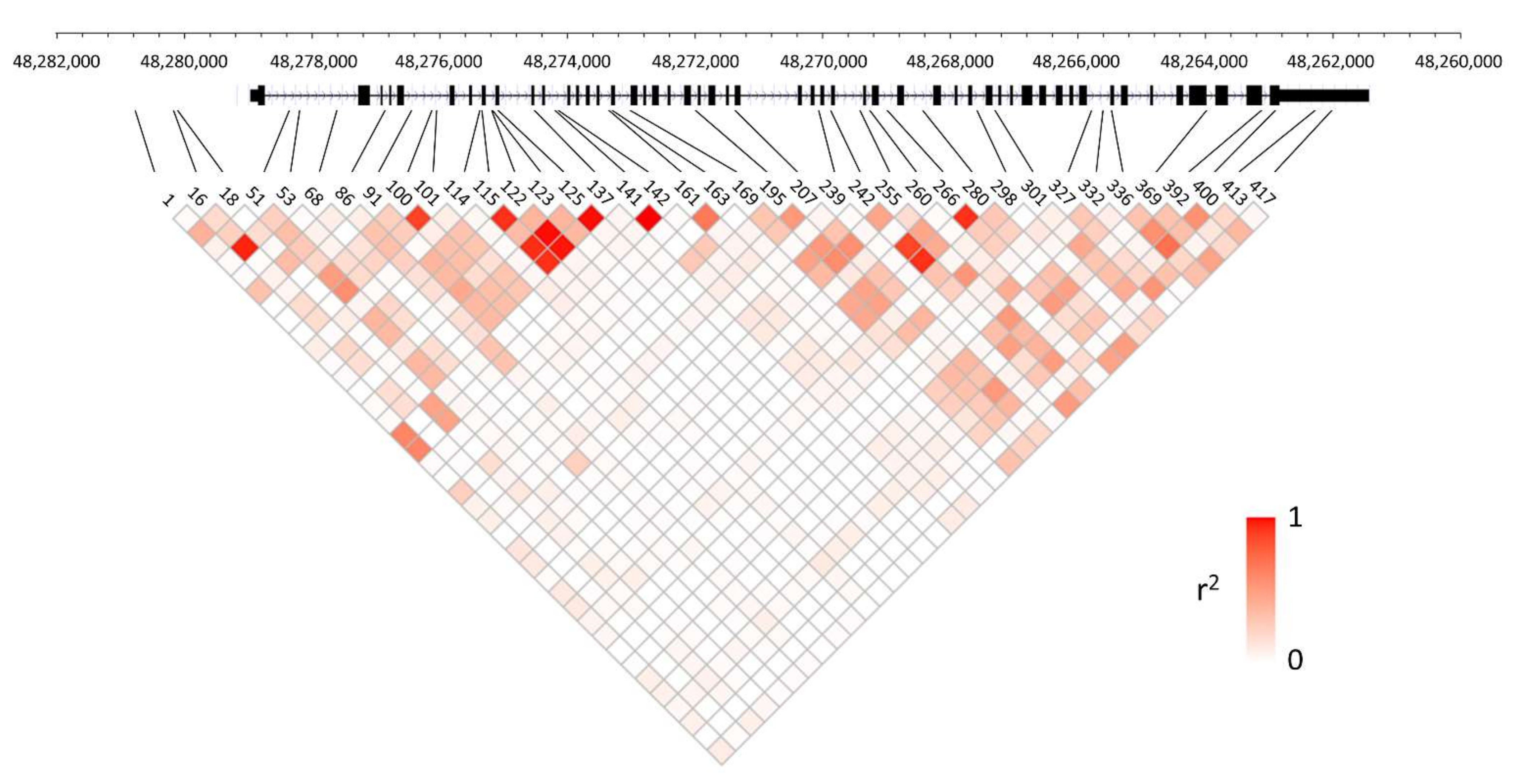
3.3. Population Patterns of Human Intron Variation
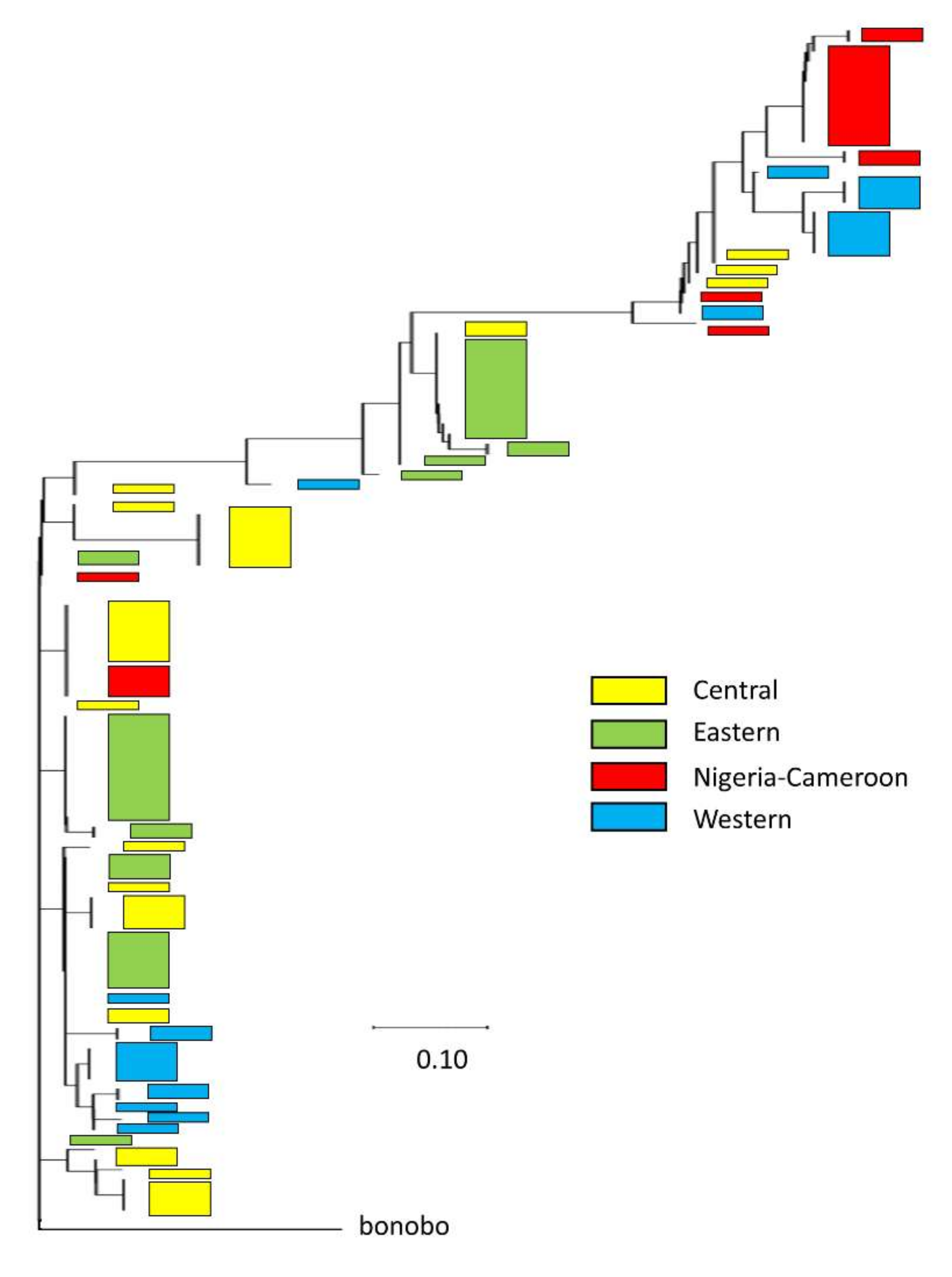
3.4. Population Patterns of Chimpanzee Intron Variation
4. Discussion
4.1. COL1A1 Protein Variation Is Higher Than Expected
4.2. Signatures of Adaptive COL1A1 Intronic Variation within Humans and Chimpanzees
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stoll, C.; Dott, B.; Roth, M.P.; Alembik, Y. Birth Prevalence Rates of Skeletal Dysplasias. Clin. Genet. 1989, 35, 88–92. [Google Scholar] [CrossRef]
- Reginster, J.-Y.; Burlet, N. Osteoporosis: A Still Increasing Prevalence. Bone 2006, 38, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Ballane, G.; Cauley, J.A.; Luckey, M.M.; El-Hajj Fuleihan, G. Worldwide Prevalence and Incidence of Osteoporotic Vertebral Fractures. Osteoporos. Int. 2017, 28, 1531–1542. [Google Scholar] [CrossRef]
- Looker, A.C.; Sarafrazi Isfahani, N.; Fan, B.; Shepherd, J.A. Trends in Osteoporosis and Low Bone Mass in Older US Adults, 2005–2006 through 2013–2014. Osteoporos. Int. 2017, 28, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.E.; Santos, M.P.; Wright, N.C. Racial and Ethnic Disparities in Bone Health and Outcomes in the United States. J. Bone. Min. Res. 2021, 36, 1881–1905. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.; Cattani, G.; Rossini, M.; Viapiana, O.; Olivi, P.; Orsolini, G.; Bertoldo, E.; Fracassi, E.; Gatti, D.; Fassio, A. Association between Exposure to Fine Particulate Matter and Osteoporosis: A Population-Based Cohort Study. Osteoporos. Int. 2022, 33, 169–176. [Google Scholar] [CrossRef]
- Min, C.; Yoo, D.M.; Wee, J.H.; Lee, H.-J.; Choi, H.G. High-Intensity Physical Activity with High Serum Vitamin D Levels Is Associated with a Low Prevalence of Osteopenia and Osteoporosis: A Population-Based Study. Osteoporos. Int. 2021, 32, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.B.; Streeten, E.A.; Shapiro, J.R.; McBride, D.; Shuldiner, A.R.; Peyser, P.A.; Mitchell, B.D. Genetic and Environmental Influences on Bone Mineral Density in Pre- and Post-Menopausal Women. Osteoporos. Int. 2005, 16, 1849–1856. [Google Scholar] [CrossRef]
- Videman, T.; Levälahti, E.; Battié, M.C.; Simonen, R.; Vanninen, E.; Kaprio, J. Heritability of BMD of Femoral Neck and Lumbar Spine: A Multivariate Twin Study of Finnish Men. J. Bone Miner. Res. 2007, 22, 1455–1462. [Google Scholar] [CrossRef]
- Liu, C.-T.; Karasik, D.; Zhou, Y.; Hsu, Y.-H.; Genant, H.K.; Broe, K.E.; Lang, T.F.; Samelson, E.J.; Demissie, S.; Bouxsein, M.L.; et al. Heritability of Prevalent Vertebral Fracture and Volumetric Bone Mineral Density and Geometry at the Lumbar Spine in Three Generations of the Framingham Study. J. Bone Miner. Res. 2012, 27, 954–958. [Google Scholar] [CrossRef]
- Estrada, K.; Styrkarsdottir, U.; Evangelou, E.; Hsu, Y.-H.; Duncan, E.L.; Ntzani, E.E.; Oei, L.; Albagha, O.M.E.; Amin, N.; Kemp, J.P.; et al. Genome-Wide Meta-Analysis Identifies 56 Bone Mineral Density Loci and Reveals 14 Loci Associated with Risk of Fracture. Nat. Genet. 2012, 44, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Huang, S.; Hou, W.; Liu, Y.; Fan, Q.; He, A.; Wen, Y.; Hao, J.; Guo, X.; Zhang, F. Integrative Analysis of GWAS, EQTLs and MeQTLs Data Suggests That Multiple Gene Sets Are Associated with Bone Mineral Density. Bone Joint Res. 2017, 6, 572–576. [Google Scholar] [CrossRef]
- Kim, S.K. Identification of 613 New Loci Associated with Heel Bone Mineral Density and a Polygenic Risk Score for Bone Mineral Density, Osteoporosis and Fracture. PLoS ONE 2018, 13, e0200785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Liu, Y.; Su, K.-J.; Greenbaum, J.; Bai, Y.; Tian, Q.; Zhao, L.-J.; Deng, H.-W.; Shen, H. A Transcriptome-Wide Association Study to Detect Novel Genes for Volumetric Bone Mineral Density. Bone 2021, 153, 116106. [Google Scholar] [CrossRef]
- Karasik, D.; Rivadeneira, F.; Johnson, M.L. The Genetics of Bone Mass and Susceptibility to Bone Diseases. Nat. Rev. Rheumatol. 2016, 12, 323–334. [Google Scholar] [CrossRef]
- Formosa, M.M.; Bergen, D.J.M.; Gregson, C.L.; Maurizi, A.; Kämpe, A.; Garcia-Giralt, N.; Zhou, W.; Grinberg, D.; Ovejero Crespo, D.; Zillikens, M.C.; et al. A Roadmap to Gene Discoveries and Novel Therapies in Monogenic Low and High Bone Mass Disorders. Front. Endocrinol. 2021, 12, 709711. [Google Scholar] [CrossRef]
- Nowlan, N.C.; Jepsen, K.J.; Morgan, E.F. Smaller, Weaker, and Less Stiff Bones Evolve from Changes in Subsistence Strategy. Osteoporos. Int. 2011, 22, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Kralick, A.E.; Zemel, B.S. Evolutionary Perspectives on the Developing Skeleton and Implications for Lifelong Health. Front. Endocrinol. 2020, 11, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-T.; Estrada, K.; Yerges-Armstrong, L.M.; Amin, N.; Evangelou, E.; Li, G.; Minster, R.L.; Carless, M.A.; Kammerer, C.M.; Oei, L.; et al. Assessment of Gene-by-Sex Interaction Effect on Bone Mineral Density. J. Bone Miner. Res. 2012, 27, 2051–2064. [Google Scholar] [CrossRef]
- Yau, M.S.; Kuipers, A.L.; Price, R.; Nicolas, A.; Tajuddin, S.M.; Handelman, S.K.; Arbeeva, L.; Chesi, A.; Hsu, Y.-H.; Liu, C.-T.; et al. A Meta-Analysis of the Transferability of Bone Mineral Density Genetic Loci Associations from European to African Ancestry Populations. J. Bone Miner. Res. 2021, 36, 469–479. [Google Scholar] [CrossRef]
- Agarwal, S.C. What Is Normal Bone Health? A Bioarchaeological Perspective on Meaningful Measures and Interpretations of Bone Strength, Loss, and Aging. Am. J. Hum. Biol. 2021, 33, e23647. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gómez, C.; Chesi, A.; Heppe, D.H.M.; Zemel, B.S.; Yin, J.-L.; Kalkwarf, H.J.; Hofman, A.; Lappe, J.M.; Kelly, A.; Kayser, M.; et al. BMD Loci Contribute to Ethnic and Developmental Differences in Skeletal Fragility across Populations: Assessment of Evolutionary Selection Pressures. Mol. Biol. Evol. 2015, 32, 2961–2972. [Google Scholar] [CrossRef] [Green Version]
- Black, A.; Tilmont, E.M.; Handy, A.M.; Scott, W.W.; Shapses, S.A.; Ingram, D.K.; Roth, G.S.; Lane, M.A. A Nonhuman Primate Model of Age-Related Bone Loss: A Longitudinal Study in Male and Premenopausal Female Rhesus Monkeys. Bone 2001, 28, 295–302. [Google Scholar] [CrossRef]
- Lipkin, E.W.; Aumann, C.A.; Newell-Morris, L.L. Evidence for Common Controls over Inheritance of Bone Quantity and Body Size from Segregation Analysis in a Pedigreed Colony of Nonhuman Primates (Macaca Nemestrina). Bone 2001, 29, 249–257. [Google Scholar] [CrossRef]
- Havill, L.M.; Allen, M.R.; Harris, J.a.K.; Levine, S.M.; Coan, H.B.; Mahaney, M.C.; Nicolella, D.P. Intracortical Bone Remodeling Variation Shows Strong Genetic Effects. Calcif. Tissue Int. 2013, 93, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Filipski, A.; Swarna, V.; Walker, A.; Hedges, S.B. Placing Confidence Limits on the Molecular Age of the Human-Chimpanzee Divergence. Proc. Natl. Acad. Sci. USA 2005, 102, 18842–18847. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, Y.; Udono, T.; Hamada, Y. Bone Mineral Density in Chimpanzees, Humans, and Japanese Macaques. Primates 2003, 44, 151–155. [Google Scholar] [CrossRef]
- Matsumura, A.; Gunji, H.; Takahashi, Y.; Nishida, T.; Okada, M. Cross-Sectional Morphology of the Femoral Neck of Wild Chimpanzees. Int. J. Primatol. 2010, 31, 219–238. [Google Scholar] [CrossRef]
- Mulhern, D.M.; Ubelaker, D.H. Bone Microstructure in Juvenile Chimpanzees. Am. J. Phys. Anthropol. 2009, 140, 368–375. [Google Scholar] [CrossRef]
- Zhu, X.; Bai, W.; Zheng, H. Twelve Years of GWAS Discoveries for Osteoporosis and Related Traits: Advances, Challenges and Applications. Bone Res. 2021, 9, 23. [Google Scholar] [CrossRef]
- Viguet-Carrin, S.; Garnero, P.; Delmas, P.D. The Role of Collagen in Bone Strength. Osteoporos. Int. 2006, 17, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, R. The Human Type I Collagen Mutation Database. Nucleic Acids Res. 1997, 25, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Marini, J.C.; Forlino, A.; Cabral, W.A.; Barnes, A.M.; San Antonio, J.D.; Milgrom, S.; Hyland, J.C.; Körkkö, J.; Prockop, D.J.; de Paepe, A.; et al. Consortium for Osteogenesis Imperfecta Mutations in the Helical Domain of Type I Collagen: Regions Rich in Lethal Mutations Align with Collagen Binding Sites for Integrins and Proteoglycans. Hum. Mutat. 2007, 28, 209–221. [Google Scholar] [CrossRef]
- Garcia-Giralt, N.; Nogués, X.; Enjuanes, A.; Puig, J.; Mellibovsky, L.; Bay-Jensen, A.; Carreras, R.; Balcells, S.; Díez-Pérez, A.; Grinberg, D. Two New Single-Nucleotide Polymorphisms in the COL1A1 Upstream Regulatory Region and Their Relationship to Bone Mineral Density. J. Bone Miner. Res. 2002, 17, 384–393. [Google Scholar] [CrossRef]
- Stewart, T.L.; Jin, H.; McGuigan, F.E.A.; Albagha, O.M.E.; Garcia-Giralt, N.; Bassiti, A.; Grinberg, D.; Balcells, S.; Reid, D.M.; Ralston, S.H. Haplotypes Defined by Promoter and Intron 1 Polymorphisms of the COLIA1 Gene Regulate Bone Mineral Density in Women. J. Clin. Endocrinol. Metab. 2006, 91, 3575–3583. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Lei, S.-F.; Xiao, S.-M.; Chen, Y.; Sun, X.; Yang, F.; Li, L.-M.; Wu, S.; Deng, H.-W. Association and Linkage Analysis of COL1A1 and AHSG Gene Polymorphisms with Femoral Neck Bone Geometric Parameters in Both Caucasian and Chinese Nuclear Families. Acta Pharmacol. Sin. 2007, 28, 375–381. [Google Scholar] [CrossRef]
- Stover, D.A.; Verrelli, B.C. Comparative Vertebrate Evolutionary Analyses of Type I Collagen: Potential of COL1a1 Gene Structure and Intron Variation for Common Bone-Related Diseases. Mol. Biol. Evol. 2011, 28, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next Generation in Gene Variant Databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.M.; Atkinson, M.; Willing, M.C.; Wallis, G.; Byers, P.H. Deletions and Duplications of Gly-Xaa-Yaa Triplet Repeats in the Triple Helical Domains of Type I Collagen Chains Disrupt Helix Formation and Result in Several Types of Osteogenesis Imperfecta. Hum. Mutat. 2001, 18, 319–326. [Google Scholar] [CrossRef]
- Cabral, W.A.; Mertts, M.V.; Makareeva, E.; Colige, A.; Tekin, M.; Pandya, A.; Leikin, S.; Marini, J.C. Type I Collagen Triplet Duplication Mutation in Lethal Osteogenesis Imperfecta Shifts Register of α Chains throughout the Helix and Disrupts Incorporation of Mutant Helices into Fibrils and Extracellular Matrix. J. Biol. Chem. 2003, 278, 10006–10012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodian, D.L.; Chan, T.-F.; Poon, A.; Schwarze, U.; Yang, K.; Byers, P.H.; Kwok, P.-Y.; Klein, T.E. Mutation and Polymorphism Spectrum in Osteogenesis Imperfecta Type II: Implications for Genotype-Phenotype Relationships. Hum. Mol. Genet. 2009, 18, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatter, D.A.; Farndale, R.W. Structural Constraints on the Evolution of the Collagen Fibril: Convergence on a 1014-Residue COL Domain. Open Biol. 2015, 5, 140220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinnijenhuis, A.J. Visualization of Genetic Drift Processes Using the Conserved Collagen 1α1 GXY Domain. J. Mol. Evol. 2019, 87, 106–130. [Google Scholar] [CrossRef]
- Boot-Handford, R.P.; Tuckwell, D.S. Fibrillar Collagen: The Key to Vertebrate Evolution? A Tale of Molecular Incest. Bioessays 2003, 25, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Aouacheria, A.; Cluzel, C.; Lethias, C.; Gouy, M.; Garrone, R.; Exposito, J.-Y. Invertebrate Data Predict an Early Emergence of Vertebrate Fibrillar Collagen Clades and an Anti-Incest Model. J. Biol. Chem. 2004, 279, 47711–47719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, H.; Okuyama, M.; Satoh, N.; Zhang, S. Molecular Evolution of Fibrillar Collagen in Chordates, with Implications for the Evolution of Vertebrate Skeletons and Chordate Phylogeny. Evol. Dev. 2006, 8, 370–377. [Google Scholar] [CrossRef]
- Morgan, C.C.; Loughran, N.B.; Walsh, T.A.; Harrison, A.J.; O’Connell, M.J. Positive Selection Neighboring Functionally Essential Sites and Disease-Implicated Regions of Mammalian Reproductive Proteins. BMC Evol. Biol. 2010, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelse, K.; Pöschl, E.; Aigner, T. Collagens—Structure, Function, and Biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, K.A.; Gallant-Behm, C.L.; Kydd, A.S.; Hart, D.A. The Basics of Soft Tissue Healing and General Factors That Influence Such Healing. Sports Med. Arthrosc. Rev. 2005, 13, 136–144. [Google Scholar] [CrossRef]
- Bornstein, P.; McKay, J.; Morishima, J.K.; Devarayalu, S.; Gelinas, R.E. Regulatory Elements in the First Intron Contribute to Transcriptional Control of the Human α 1(I) Collagen Gene. Proc. Natl. Acad. Sci. USA 1987, 84, 8869–8873. [Google Scholar] [CrossRef] [Green Version]
- Mann, V.; Hobson, E.E.; Li, B.; Stewart, T.L.; Grant, S.F.; Robins, S.P.; Aspden, R.M.; Ralston, S.H. A COL1A1 Sp1 Binding Site Polymorphism Predisposes to Osteoporotic Fracture by Affecting Bone Density and Quality. J. Clin. Investig. 2001, 107, 899–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; van’t Hof, R.J.; Albagha, O.M.E.; Ralston, S.H. Promoter and Intron 1 Polymorphisms of COL1A1 Interact to Regulate Transcription and Susceptibility to Osteoporosis. Hum. Mol. Genet. 2009, 18, 2729–2738. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, H.; Chen, K.; Wu, B.; Liu, H. Association of Polymorphisms Rs1800012 in COL1A1 with Sports-Related Tendon and Ligament Injuries: A Meta-Analysis. Oncotarget 2017, 8, 27627–27634. [Google Scholar] [CrossRef] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of Nonneutral Substitution Rates on Mammalian Phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.-F.; Poon, A.; Basu, A.; Addleman, N.R.; Chen, J.; Phong, A.; Byers, P.H.; Klein, T.E.; Kwok, P.-Y. Natural Variation in Four Human Collagen Genes across an Ethnically Diverse Population. Genomics 2008, 91, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O.; Abel, H.J.; Regier, A.A.; Corvelo, A.; Clarke, W.E.; Musunuri, R.; Nagulapalli, K.; et al. High coverage whole genome sequencing of the expanded 1000 genomes project cohort including 602 trios. bioRxiv 2021. [Google Scholar] [CrossRef]
- Keinan, A.; Clark, A.G. Recent Explosive Human Population Growth Has Resulted in an Excess of Rare Genetic Variants. Science 2012, 336, 740–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tishkoff, S.A.; Verrelli, B.C. Patterns of Human Genetic Diversity: Implications for Human Evolutionary History and Disease. Annu. Rev. Genom. Hum. Genet. 2003, 4, 293–340. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.C.; Tishkoff, S.A. African Genetic Diversity: Implications for Human Demographic History, Modern Human Origins, and Complex Disease Mapping. Annu. Rev. Genom. Hum. Genet. 2008, 9, 403–433. [Google Scholar] [CrossRef] [Green Version]
- Henn, B.M.; Gignoux, C.R.; Jobin, M.; Granka, J.M.; Macpherson, J.M.; Kidd, J.M.; Rodríguez-Botigué, L.; Ramachandran, S.; Hon, L.; Brisbin, A.; et al. Hunter-Gatherer Genomic Diversity Suggests a Southern African Origin for Modern Humans. Proc. Natl. Acad. Sci. USA 2011, 108, 5154–5162. [Google Scholar] [CrossRef] [Green Version]
- Basel, D.; Steiner, R.D. Osteogenesis Imperfecta: Recent Findings Shed New Light on This Once Well-Understood Condition. Genet. Med. 2009, 11, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Prado-Martinez, J.; Sudmant, P.H.; Kidd, J.M.; Li, H.; Kelley, J.L.; Lorente-Galdos, B.; Veeramah, K.R.; Woerner, A.E.; O’Connor, T.D.; Santpere, G.; et al. Great Ape Genetic Diversity and Population History. Nature 2013, 499, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Manuel, M.; Kuhlwilm, M.; Frandsen, P.; Sousa, V.C.; Desai, T.; Prado-Martinez, J.; Hernandez-Rodriguez, J.; Dupanloup, I.; Lao, O.; Hallast, P.; et al. Chimpanzee Genomic Diversity Reveals Ancient Admixture with Bonobos. Science 2016, 354, 477–481. [Google Scholar] [CrossRef]
- Won, Y.-J.; Hey, J. Divergence Population Genetics of Chimpanzees. Mol. Biol. Evol. 2005, 22, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becquet, C.; Przeworski, M. A New Approach to Estimate Parameters of Speciation Models with Application to Apes. Genome Res. 2007, 17, 1505–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, A.C.; Griffiths, R.C.; Zegura, S.L.; Hammer, M.F. High Levels of Y-Chromosome Nucleotide Diversity in the Genus Pan. Proc. Natl. Acad. Sci. USA 2002, 99, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Wiebe, V.; Pääbo, S.; Przeworski, M. Evidence for a Complex Demographic History of Chimpanzees. Mol. Biol. Evol. 2004, 21, 799–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claw, K.G.; Tito, R.Y.; Stone, A.C.; Verrelli, B.C. Haplotype Structure and Divergence at Human and Chimpanzee Serotonin Transporter and Receptor Genes: Implications for Behavioral Disorder Association Analyses. Mol. Biol. Evol. 2010, 27, 1518–1529. [Google Scholar] [CrossRef] [Green Version]
- Verrelli, B.C.; Tishkoff, S.A.; Stone, A.C.; Touchman, J.W. Contrasting Histories of G6PD Molecular Evolution and Malarial Resistance in Humans and Chimpanzees. Mol. Biol. Evol. 2006, 23, 1592–1601. [Google Scholar] [CrossRef] [Green Version]
- Verrelli, B.C.; Lewis, C.M.; Stone, A.C.; Perry, G.H. Different Selective Pressures Shape the Molecular Evolution of Color Vision in Chimpanzee and Human Populations. Mol. Biol. Evol. 2008, 25, 2735–2743. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, R.M.; Haussler, D.; Kent, W.J. The UCSC Genome Browser and Associated Tools. Brief Bioinform. 2013, 14, 144–161. [Google Scholar] [CrossRef] [Green Version]
- Hothorn, T.; Hornik, K.; van de Wiel, M.A.; Zeileis, A. Implementing a Class of Permutation Tests: The coin Package. J. Stat. Soft. 2008, 28, 1–23. [Google Scholar] [CrossRef]
- McDonald, J.H.; Kreitman, M. Adaptive Protein Evolution at the Adh Locus in Drosophila. Nature 1991, 351, 652–654. [Google Scholar] [CrossRef]
- Watterson, G.A. On the Number of Segregating Sites in Genetical Models without Recombination. Theor. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Hudson, R.R.; Slatkin, M.; Maddison, W.P. Estimation of Levels of Gene Flow from DNA Sequence Data. Genetics 1992, 132, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.R. A New Statistic for Detecting Genetic Differentiation. Genetics 2000, 155, 2011–2014. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. 1000 Genomes Project Analysis Group. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Smith, N.J.; Donnelly, P. A New Statistical Method for Haplotype Reconstruction from Population Data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Sałacińska, K.; Pinkier, I.; Rutkowska, L.; Chlebna-Sokół, D.; Jakubowska-Pietkiewicz, E.; Michałus, I.; Kępczyński, Ł.; Salachna, D.; Jamsheer, A.; Bukowska-Olech, E.; et al. Novel Mutations Within Collagen Alpha1(I) and Alpha2(I) Ligand-Binding Sites, Broadening the Spectrum of Osteogenesis Imperfecta—Current Insights into Collagen Type I Lethal Regions. Front. Genet. 2021, 12, 692978. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T. The Nearly Neutral Theory of Molecular Evolution. Annu. Rev. Ecol. Syst. 1992, 23, 263–286. [Google Scholar] [CrossRef]
- Charlesworth, J.; Eyre-Walker, A. The McDonald-Kreitman Test and Slightly Deleterious Mutations. Mol. Biol. Evol. 2008, 25, 1007–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housman, G.; Briscoe, E.; Gilad, Y. Evolutionary insights into primate skeletal gene regulation using a comparative cell culture model. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bradnam, K.R.; Korf, I. Longer First Introns Are a General Property of Eukaryotic Gene Structure. PLoS ONE 2008, 3, e3093. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A.; Pritchard, J.K.; Weber, J.L.; Cann, H.M.; Kidd, K.K.; Zhivotovsky, L.A.; Feldman, M.W. Genetic Structure of Human Populations. Science 2002, 298, 2381–2385. [Google Scholar] [CrossRef] [Green Version]
- Tishkoff, S.A.; Reed, F.A.; Friedlaender, F.R.; Ehret, C.; Ranciaro, A.; Froment, A.; Hirbo, J.B.; Awomoyi, A.A.; Bodo, J.-M.; Doumbo, O.; et al. The Genetic Structure and History of Africans and African Americans. Science 2009, 324, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tishkoff, S.A.; Verrelli, B.C. Role of Evolutionary History on Haplotype Block Structure in the Human Genome: Implications for Disease Mapping. Curr. Opin. Genet. Dev. 2003, 13, 569–575. [Google Scholar] [CrossRef]
- Thomson, R.; Pritchard, J.K.; Shen, P.; Oefner, P.J.; Feldman, M.W. Recent Common Ancestry of Human Y Chromosomes: Evidence from DNA Sequence Data. Proc. Natl. Acad. Sci. USA 2000, 97, 7360–7365. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, R.C.; Tavaré, S. The Age of a Mutation in a General Coalescent Tree. Commun. Statistics. Stoch. Models 1998, 14, 273–295. [Google Scholar] [CrossRef]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6,515 Exomes Reveals the Recent Origin of Most Human Protein-Coding Variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Gonder, M.K.; Locatelli, S.; Ghobrial, L.; Mitchell, M.W.; Kujawski, J.T.; Lankester, F.J.; Stewart, C.-B.; Tishkoff, S.A. Evidence from Cameroon Reveals Differences in the Genetic Structure and Histories of Chimpanzee Populations. Proc. Natl. Acad. Sci. USA 2011, 108, 4766–4771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyko, A.R.; Williamson, S.H.; Indap, A.R.; Degenhardt, J.D.; Hernandez, R.D.; Lohmueller, K.E.; Adams, M.D.; Schmidt, S.; Sninsky, J.J.; Sunyaev, S.R.; et al. Assessing the Evolutionary Impact of Amino Acid Mutations in the Human Genome. PLoS Genet. 2008, 4, e1000083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohmueller, K.E.; Indap, A.R.; Schmidt, S.; Boyko, A.R.; Hernandez, R.D.; Hubisz, M.J.; Sninsky, J.J.; White, T.J.; Sunyaev, S.R.; Nielsen, R.; et al. Proportionally More Deleterious Genetic Variation in European than in African Populations. Nature 2008, 451, 994–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, C.D.; Fledel-Alon, A.; Williamson, S.; Nielsen, R.; Hubisz, M.T.; Glanowski, S.; Tanenbaum, D.M.; White, T.J.; Sninsky, J.J.; Hernandez, R.D.; et al. Natural Selection on Protein-Coding Genes in the Human Genome. Nature 2005, 437, 1153–1157. [Google Scholar] [CrossRef]
- Anderson-Trocmé, L.; Farouni, R.; Bourgey, M.; Kamatani, Y.; Higasa, K.; Seo, J.-S.; Kim, C.; Matsuda, F.; Gravel, S. Legacy Data Confound Genomics Studies. Mol. Biol. Evol. 2020, 37, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Belsare, S.; Levy-Sakin, M.; Mostovoy, Y.; Durinck, S.; Chaudhuri, S.; Xiao, M.; Peterson, A.S.; Kwok, P.-Y.; Seshagiri, S.; Wall, J.D. Evaluating the Quality of the 1000 Genomes Project Data. BMC Genom. 2019, 20, 620. [Google Scholar] [CrossRef] [Green Version]
- Rensberger, J.M.; Watabe, M. Fine Structure of Bone in Dinosaurs, Birds and Mammals. Nature 2000, 406, 619–622. [Google Scholar] [CrossRef]
- Abbott, S.; Trinkaus, E.; Burr, D.B. Dynamic Bone Remodeling in Later Pleistocene Fossil Hominids. Am. J. Phys. Anthropol. 1996, 99, 585–601. [Google Scholar] [CrossRef]
- Larsen, C.S. Biological Changes in Human Populations with Agriculture. Annu. Rev. Anthropol. 1995, 24, 185–213. [Google Scholar] [CrossRef]
- Przeworski, M.; Coop, G.; Wall, J.D. The Signature of Positive Selection on Standing Genetic Variation. Evolution 2005, 59, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Bitarello, B.D.; de Filippo, C.; Teixeira, J.C.; Schmidt, J.M.; Kleinert, P.; Meyer, D.; Andrés, A.M. Signatures of Long-Term Balancing Selection in Human Genomes. Genome Biol. Evol. 2018, 10, 939–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ram, O.; Goren, A.; Amit, I.; Shoresh, N.; Yosef, N.; Ernst, J.; Kellis, M.; Gymrek, M.; Issner, R.; Coyne, M.; et al. Combinatorial Patterning of Chromatin Regulators Uncovered by Genome-Wide Location Analysis in Human Cells. Cell 2011, 147, 1628–1639. [Google Scholar] [CrossRef] [Green Version]
- Verrelli, B.C.; McDonald, J.H.; Argyropoulos, G.; Destro-Bisol, G.; Froment, A.; Drousiotou, A.; Lefranc, G.; Helal, A.N.; Loiselet, J.; Tishkoff, S.A. Evidence for Balancing Selection from Nucleotide Sequence Analyses of Human G6PD. Am. J. Hum. Genet. 2002, 71, 1112–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrelli, B.C.; Tishkoff, S.A. Signatures of Selection and Gene Conversion Associated with Human Color Vision Variation. Am. J. Hum. Genet. 2004, 75, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Tishkoff, S.A.; Reed, F.A.; Ranciaro, A.; Voight, B.F.; Babbitt, C.C.; Silverman, J.S.; Powell, K.; Mortensen, H.M.; Hirbo, J.B.; Osman, M.; et al. Convergent Adaptation of Human Lactase Persistence in Africa and Europe. Nat. Genet. 2007, 39, 31–40. [Google Scholar] [CrossRef]
- Gurdasani, D.; Carstensen, T.; Fatumo, S.; Chen, G.; Franklin, C.S.; Prado-Martinez, J.; Bouman, H.; Abascal, F.; Haber, M.; Tachmazidou, I.; et al. Uganda Genome Resource Enables Insights into Population History and Genomic Discovery in Africa. Cell 2019, 179, 984–1002.e36. [Google Scholar] [CrossRef]
- Hancock, A.M.; Witonsky, D.B.; Ehler, E.; Alkorta-Aranburu, G.; Beall, C.; Gebremedhin, A.; Sukernik, R.; Utermann, G.; Pritchard, J.; Coop, G.; et al. Colloquium Paper: Human Adaptations to Diet, Subsistence, and Ecoregion Are Due to Subtle Shifts in Allele Frequency. Proc. Natl. Acad. Sci. USA 2010, 107, 8924–8930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.H.; Seet, S.H.; Bascom, C.C.; Isfort, R.J.; Bard, F. Red-COLA1: A Human Fibroblast Reporter Cell Line for Type I Collagen Transcription. Sci. Rep. 2020, 10, 19723. [Google Scholar] [CrossRef]
- Al-Barghouthi, B.M.; Mesner, L.D.; Calabrese, G.M.; Brooks, D.; Tommasini, S.M.; Bouxsein, M.L.; Horowitz, M.C.; Rosen, C.J.; Nguyen, K.; Haddox, S.; et al. Systems Genetics in Diversity Outbred Mice Inform BMD GWAS and Identify Determinants of Bone Strength. Nat. Commun. 2021, 12, 3408. [Google Scholar] [CrossRef]
- Stone, A.C.; Verrelli, B.C. Focusing on Comparative Ape Population Genetics in the Post-Genomic Age. Curr. Opin. Genet. Dev. 2006, 16, 586–591. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stover, D.A.; Housman, G.; Stone, A.C.; Rosenberg, M.S.; Verrelli, B.C. Evolutionary Genetic Signatures of Selection on Bone-Related Variation within Human and Chimpanzee Populations. Genes 2022, 13, 183. https://doi.org/10.3390/genes13020183
Stover DA, Housman G, Stone AC, Rosenberg MS, Verrelli BC. Evolutionary Genetic Signatures of Selection on Bone-Related Variation within Human and Chimpanzee Populations. Genes. 2022; 13(2):183. https://doi.org/10.3390/genes13020183
Chicago/Turabian StyleStover, Daryn A., Genevieve Housman, Anne C. Stone, Michael S. Rosenberg, and Brian C. Verrelli. 2022. "Evolutionary Genetic Signatures of Selection on Bone-Related Variation within Human and Chimpanzee Populations" Genes 13, no. 2: 183. https://doi.org/10.3390/genes13020183
APA StyleStover, D. A., Housman, G., Stone, A. C., Rosenberg, M. S., & Verrelli, B. C. (2022). Evolutionary Genetic Signatures of Selection on Bone-Related Variation within Human and Chimpanzee Populations. Genes, 13(2), 183. https://doi.org/10.3390/genes13020183