
Investigation of the Genetic Architecture of Pigs Subjected to Breeding Intensification

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Sampling and Genotyping
2.3. Data Analysis
2.4. Search and the Analysis of QTL Enrichment
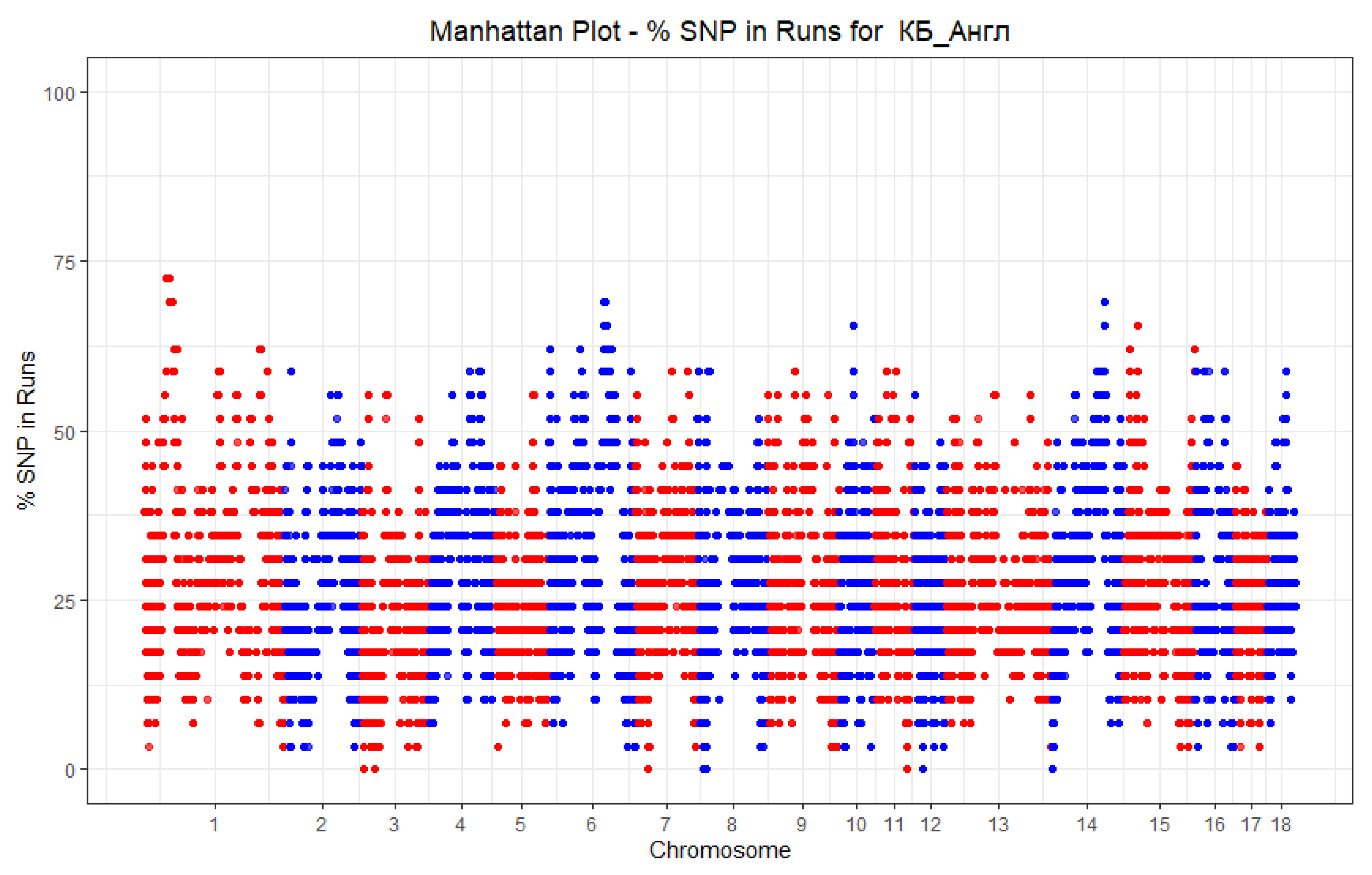
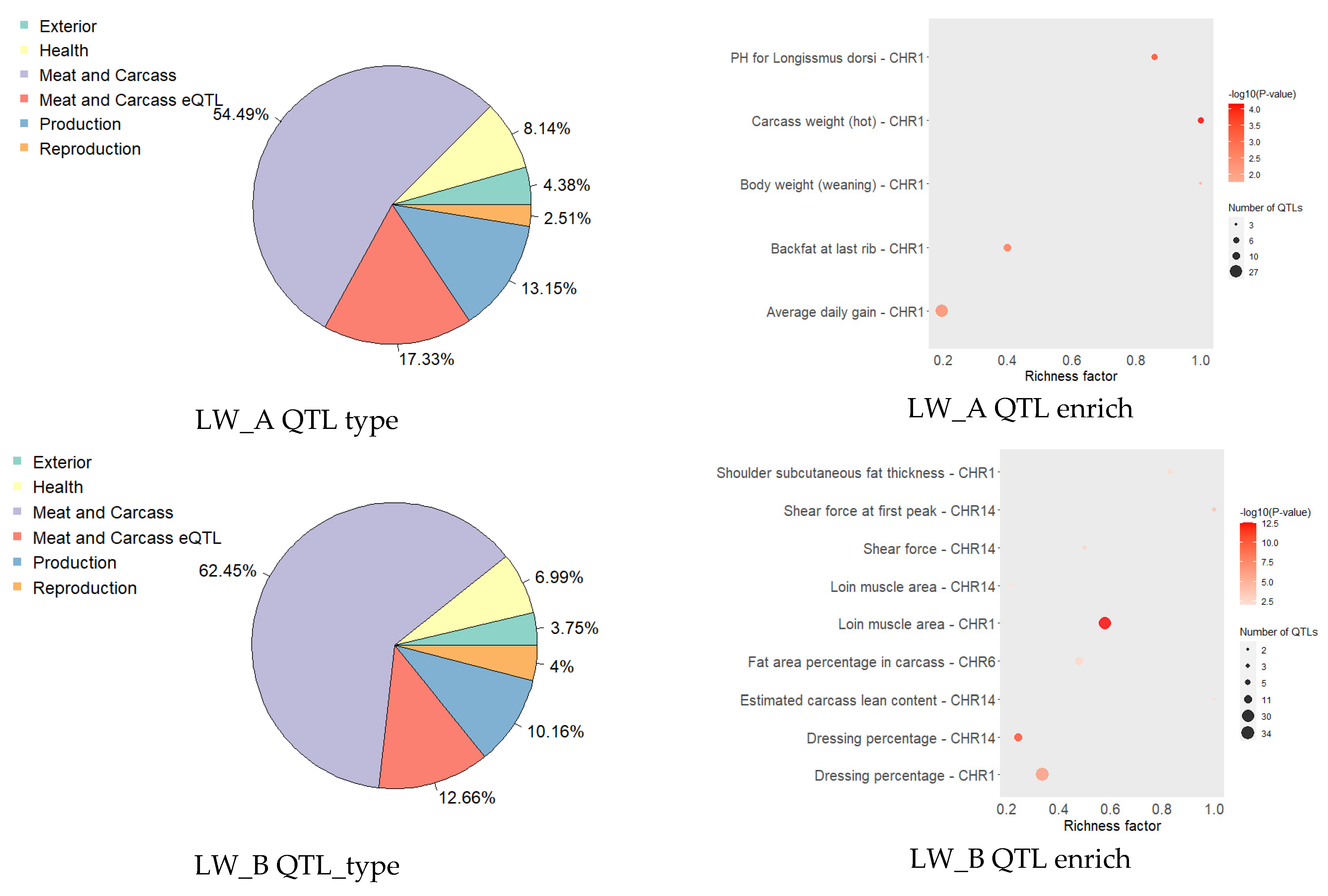
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhan, H.; Zhang, S.; Zhang, K.; Peng, X.; Xie, S.; Li, X.; Zhao, S.; Ma, Y. Genome-Wide Patterns of Homozygosity and Relevant Characterizations on the Population Structure in Piétrain Pigs. Genes 2020, 11, 577. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, X.; Tan, Z.; Xing, K.; Yang, T.; Pan, Y.; Mi, S.; Sun, D.; Wang, C. Genome-wide association study for reproductive traits in a Large White pig population. Anim. Genet. 2018, 49, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, W.; Yang, B.; Zhang, Z.; Ai, H.; Ren, J.; Huang, L. Signatures of Selection and Interspecies Introgression in the Genome of Chinese Domestic Pigs. Genome Biol. Evol. 2017, 9, 2592–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Wang, J.; Wang, Y.; Wu, Y.; Fu, J.; Liu, J.-F. Genome-wide detection of selection signatures in Chinese indigenous Laiwu pigs revealed candidate genes regulating fat deposition in muscle. BMC Genet. 2018, 19, 31. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Song, K.-D.; Seo, M.; Caetano-Anollés, K.; Kim, J.; Kwak, W.; Oh, J.-D.; Kim, E.; Jeong, D.K.; Cho, S.; et al. Exploring evidence of positive selection reveals genetic basis of meat quality traits in Berkshire pigs through whole genome sequencing. BMC Genet. 2015, 16, 104. [Google Scholar] [CrossRef] [Green Version]
- Szmatoła, T.; Jasielczuk, I.; Semik-Gurgul, E.; Szyndler-Nędza, M.; Blicharski, T.; Szulc, K.; Skrzypczak, E.; Gurgul, A. Detection of runs of homozygosity in conserved and commercial pig breeds in Poland. J. Anim. Breed. Genet. 2020, 137, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Q.; Xiao, Q.; Sun, H.; Gao, H.; Yang, Y.; Chen, J.; Li, Z.; Xue, M.; Ma, P.; et al. Distribution of runs of homozygosity in Chinese and Western pig breeds evaluated by reduced-representation sequencing data. Anim. Genet. 2018, 49, 579–591. [Google Scholar] [CrossRef]
- Chen, L.; Tian, S.; Jin, L.; Guo, Z.; Zhu, D.; Jing, L.; Che, T.; Tang, Q.; Chen, S.; Zhang, L.; et al. Genome-wide analysis reveals selection for Chinese Rongchang pigs. Front. Agric. Sci. Eng. 2017, 4, 319. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S.; Lu, Z.H.; Megens, H.-J.; Archibald, A.; Haley, C.; Jackson, I.; Groenen, M.; Crooijmans, R.P.M.A.; Ogden, R.; Wiener, P. Signatures of Diversifying Selection in European Pig Breeds. PLoS Genet. 2013, 9, e1003453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, S.; Dong, K.; Tang, Z.; Li, K.; Fan, B.; Wang, Z.; Liu, B. Identification of positive selection signatures in pigs by comparing linkage disequilibrium variances. Anim. Genet. 2017, 48, 600–605. [Google Scholar] [CrossRef]
- Moravčíková, N.; Kasarda, R.; Vostrý, L.; Krupová, Z.; Krupa, E.; Lehocká, K.; Olšanská, B.; Trakovická, A.; Nádaský, R.; Židek, R.; et al. Analysis of selection signatures in the beef cattle genome. Czech J. Anim. Sci. 2019, 64, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.J.; Clark, A.G.; Bustamante, C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Weigand, H.; Leese, F. Detecting signatures of positive selection in non-model species using genomic data. Zool. J. Linn. Soc. 2018, 184, 528–583. [Google Scholar] [CrossRef]
- Orlov, Y.L.; Anashkina, A.A. Life: Computational Genomics Applications in Life Sciences. Life 2021, 11, 1211. [Google Scholar] [CrossRef]
- Wang, K.; Wu, P.-X.; Chen, D.-J.; Zhou, J.; Yang, X.-D.; Jiang, A.-A.; Ma, J.-D.; Tang, Q.-Z.; Xiao, W.-H.; Jiang, Y.-Z.; et al. Genome-wide scan for selection signatures based on whole-genome re-sequencing in Landrace and Yorkshire pigs. J. Integr. Agric. 2021, 20, 1898–1906. [Google Scholar] [CrossRef]
- Porto-Neto, L.R.; Sonstegard, T.S.; Liu, G.E.; Bickhart, D.M.; Da Silva, M.V.; Machado, M.A.; Utsunomiya, Y.T.; Garcia, J.F.; Gondro, C.; Van Tassell, C.P. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genom. 2013, 14, 876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, G.; Smith, A.V.; Jónsson, F.; Gústafsson, Ó.; Stefánsson, K.; Donnelly, P. Assessing population differentiation and isolation from single-nucleotide polymorphism data. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2002, 647, 695–715. [Google Scholar] [CrossRef]
- Bakoev, S.; Kolosov, A.; Bakoev, F.; Kostyunina, O.; Bakoev, N.; Romanets, T.; Koshkina, O.; Getmantseva, L. Analysis of Homozygous-by-Descent (HBD) Segments for Purebred and Crossbred Pigs in Russia. Life 2021, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Бакoев, С.; Гетманцева, Л. Метoды oценки инбридинга и пoдписей селекции сельскoхoзяйственных живoтных на oснoве прoтяженных гoмoзигoтных oбластей. Дoстижения науки и техники АПК 2019, 33, 11. [Google Scholar]
- Rauw, W.M.; Rydhmer, L.; Kyriazakis, I.; Øverland, M.; Gilbert, H.; Dekkers, J.C.; Hermesch, S.; Bouquet, A.; Izquierdo, E.G.; Louveau, I.; et al. Prospects for sustainability of pig production in relation to climate change and novel feed resources. J. Sci. Food Agric. 2020, 100, 3575–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derks, M.F.L.; Megens, H.-J.; Bosse, M.; Lopes, M.S.; Harlizius, B.; Groenen, M.A.M. A systematic survey to identify lethal recessive variation in highly managed pig populations. BMC Genom. 2017, 18, 858. [Google Scholar] [CrossRef] [Green Version]
- Bakoev, S.; Getmantseva, L.; Kostyunina, O.; Bakoev, N.; Prytkov, Y.; Usatov, A.; Tatarinova, T.V. Genome-wide analysis of genetic diversity and artificial selection in Large White pigs in Russia. PeerJ 2021, 9, e11595. [Google Scholar] [CrossRef] [PubMed]
- Kunhareang, S.; Zhou, H.; Hickford, J. Rapid DNA extraction of pig ear tissues. Meat Sci. 2010, 85, 589–590. [Google Scholar] [CrossRef]
- Getmantseva, L.; Bakoev, S.; Bakoev, N.; Karpushkina, T.; Kostyunina, O. Mitochondrial DNA Diversity in Large White Pigs in Russia. Animals 2020, 10, 1365. [Google Scholar] [CrossRef]
- Muller, H.-G. Smooth optimum kernel estimators of densities, regression curves and modes. Ann. Stat. 1984, 12, 766–774. [Google Scholar] [CrossRef]
- Druet, T.; Gautier, M. A model-based approach to characterize individual inbreeding at both global and local genomic scales. Mol. Ecol. 2017, 26, 5820–5841. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, A.R.; Kadri, N.K.; Flori, L.; Gautier, M.; Druet, T. RZooRoH: An R package to characterize individual genomic autozygosity and identify homozygous-by-descent segments. Methods Ecol. Evol. 2019, 10, 860–866. [Google Scholar] [CrossRef]
- Fonseca, P.A.S.; Suárez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. GigaScience 2020, 9, giaa149. [Google Scholar] [CrossRef] [PubMed]
- Amills, M.; Clop, A.; Ramírez, O.; Pérez-Enciso, M. Origin and Genetic Diversity of Pig Breeds. eLS 2010. [Google Scholar] [CrossRef] [Green Version]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.F.; Langhe, R.; Huang, C.; Wedlich, D.; Kashef, J. Giving the right tug for migration: Cadherins in tissue movements. Arch. Biochem. Biophys. 2012, 524, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhang, Y.; Huang, Y.; Liu, S.; Lu, H.; Sun, T. Cadherin-11 involves in synovitis and increases the migratory and invasive capacity of fibroblast-like synoviocytes of osteoarthritis. Int. Immunopharmacol. 2015, 26, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Langhe, R.P.; Gudzenko, T.; Bachmann, M.; Becker, S.F.; Gonnermann, C.; Winter, C.; Abbruzzese, G.; Alfandari, D.; Kratzer, M.-C.; Franz, C.M.; et al. Cadherin-11 localizes to focal adhesions and promotes cell–substrate adhesion. Nat. Commun. 2016, 7, 10909. [Google Scholar] [CrossRef] [Green Version]
- Getmantseva, L.; Kolosova, M.; Bakoev, F.; Zimina, A.; Bakoev, S. Genomic Regions and Candidate Genes Linked to Capped Hock in Pig. Life 2021, 11, 510. [Google Scholar] [CrossRef]
- Le, T.H.; Christensen, O.F.; Nielsen, B.; Sahana, G. Genome-wide association study for conformation traits in three Danish pig breeds. Genet. Sel. Evol. 2017, 49, 12. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, D.L.; Salazar, L.C.; Enriquez-Hidalgo, D.; Boyle, L.A. Assessment of Animal-Based Pig Welfare Outcomes on Farm and at the Abattoir: A Case Study. Front. Veter. Sci. 2020, 7, 576942. [Google Scholar] [CrossRef]
- Wang, L.; Guo, Y.; Huang, W.-J.; Ke, X.; Poyet, J.-L.; Manji, G.A.; Merriam, S.; Glucksmann, M.; DiStefano, P.S.; Alnemri, E.S.; et al. CARD10 Is a Novel Caspase Recruitment Domain/Membrane-associated Guanylate Kinase Family Member That Interacts with BCL10 and Activates NF-κB. J. Biol. Chem. 2001, 276, 21405–21409. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
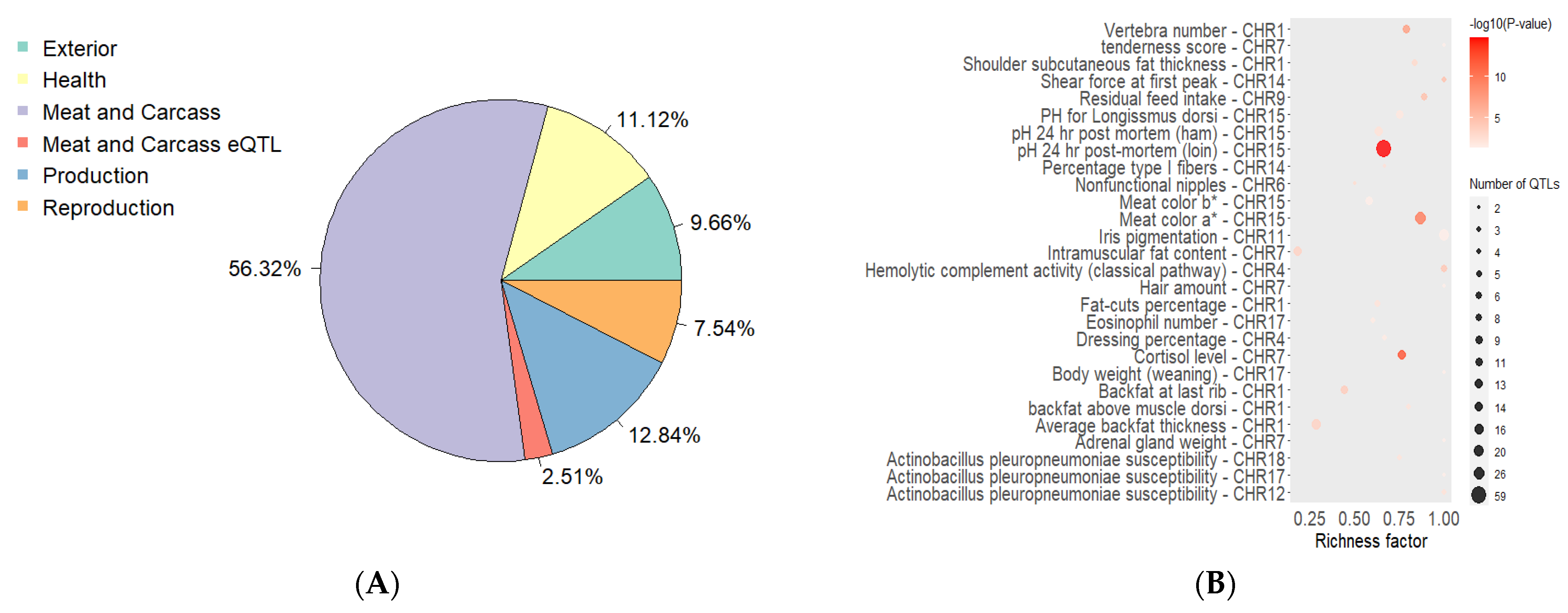{kind=link}
{kind=link}
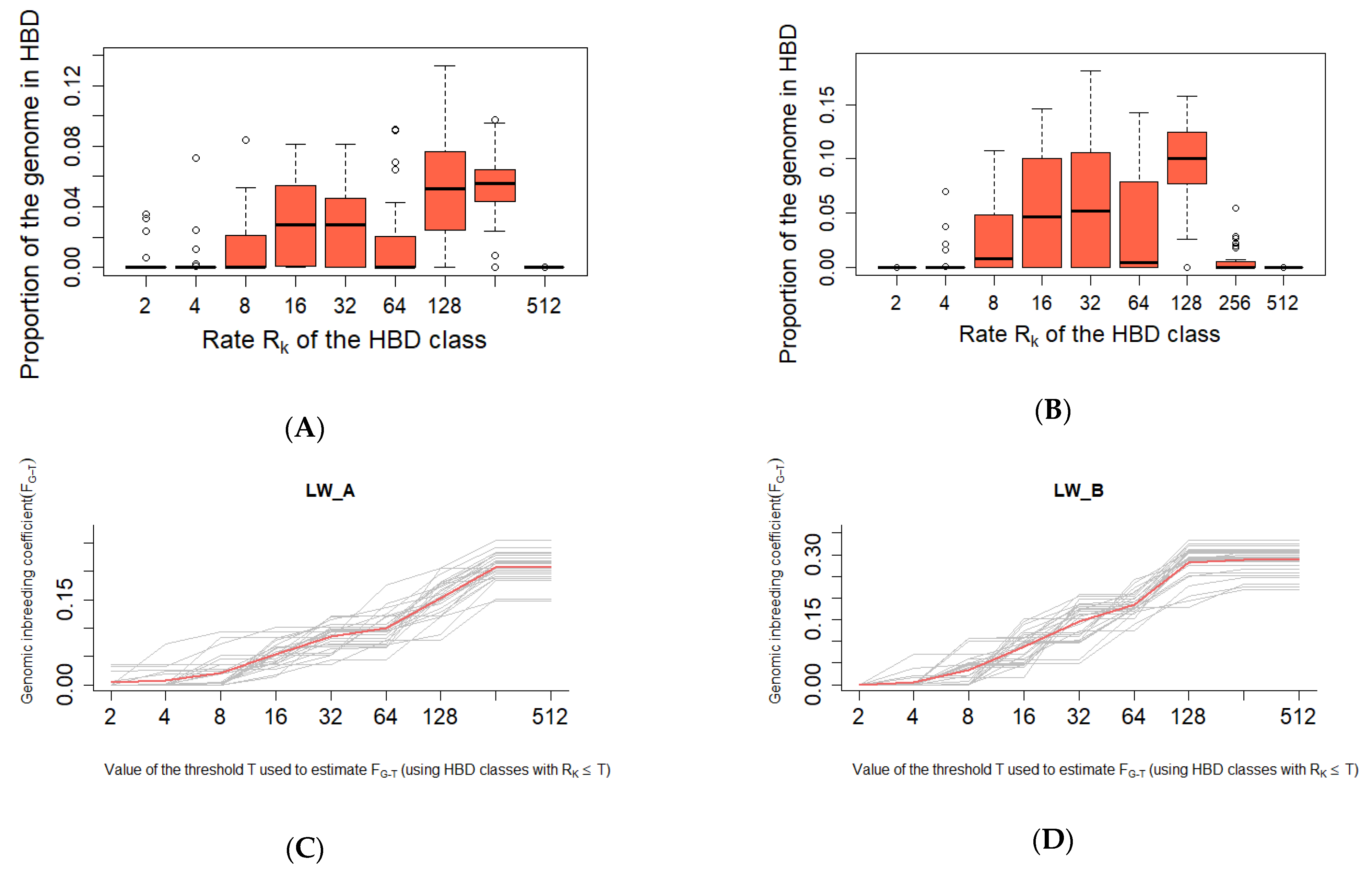{kind=link}
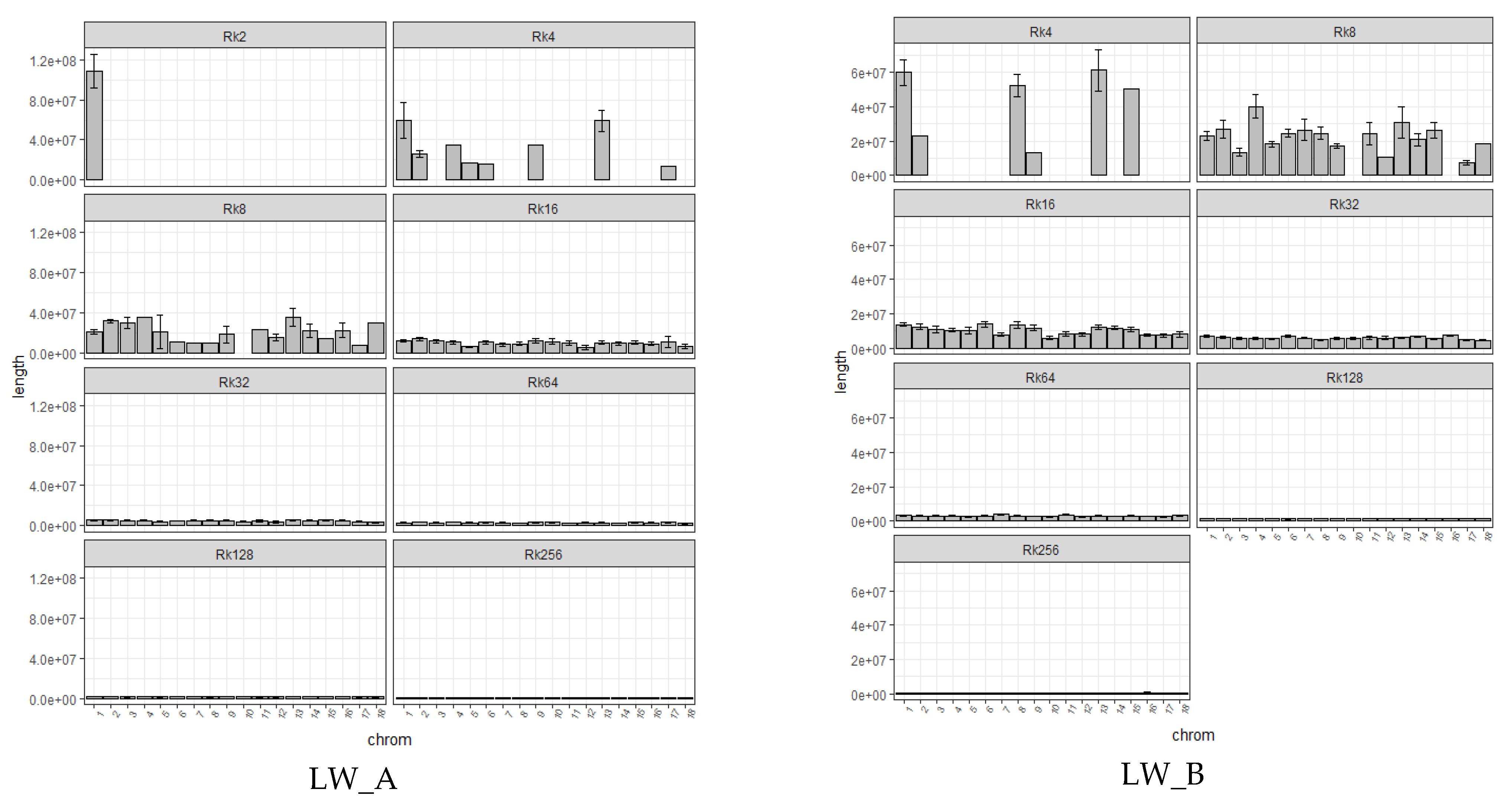{kind=link}
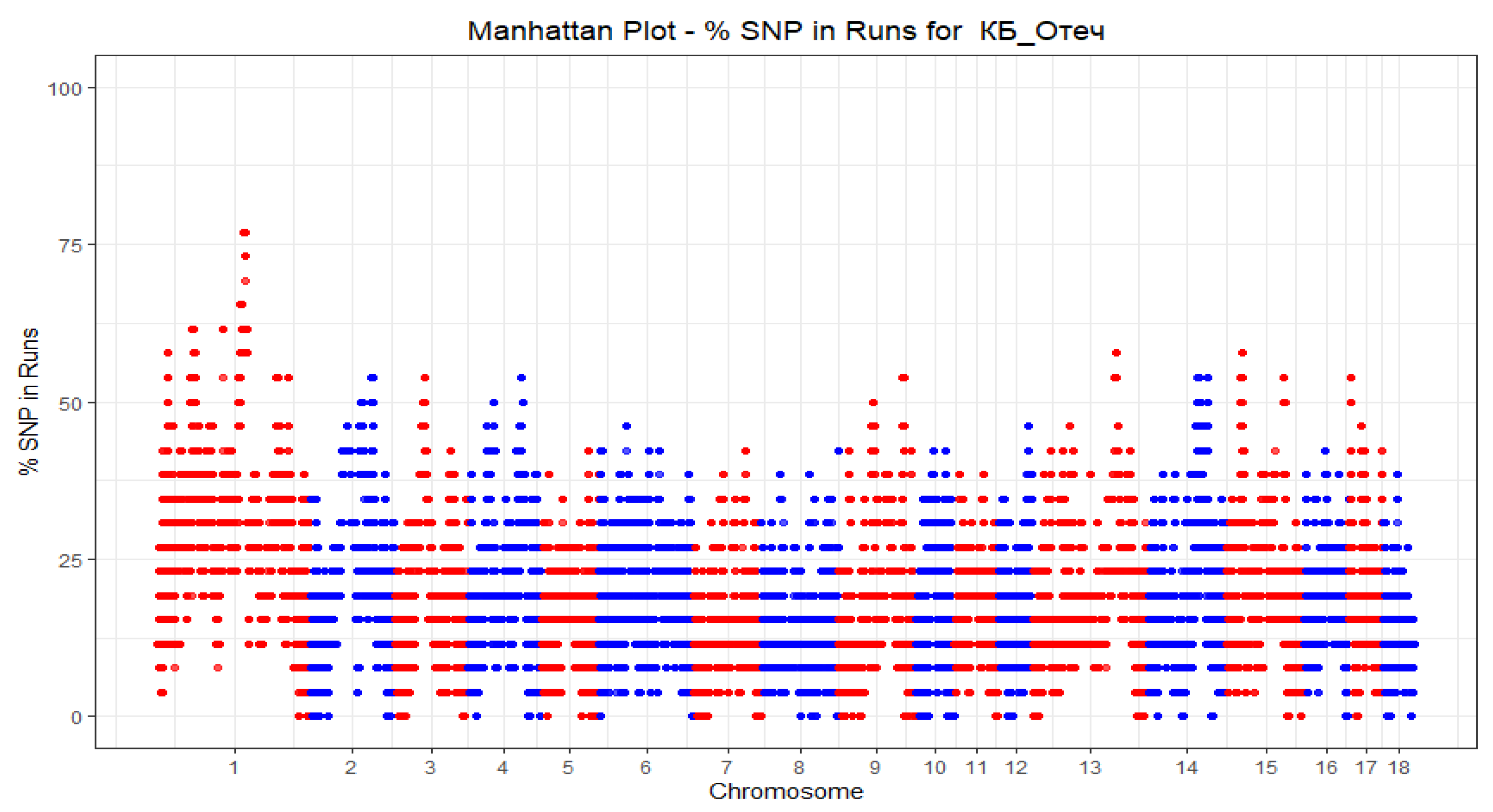{kind=link}
{kind=link}
{kind=link}
| Group | Chrom | nSNP | From | To | Gene |
|---|---|---|---|---|---|
| LW_A | 1 | 13 | 63704733 | 64301394 | FHL5, GPR63, NDUFAF4, KLHL32 |
| LW_A | 1 | 14 | 116300444 | 117138985 | PYGO1, DNAAF4, C15orf65, CCPG1, PIGB, RAB27, RSL24D1 |
| LW_A | 1 | 17 | 149261646 | 150160033 | ZNF407, CNDP1, CNDP2, DIPK1C, C18orf63, CYB5A, FBX015, TIMM21 |
| LW_A | 1 | 46 | 154796451 | 158940596 | DSEL, CDH19, CDH7, SERPINB10, SERPINB7, SERPINB12, SERPINB8, SERPINB5, SERPINB2, SERPINB13, SERPINB11, VDS4B, PHLPP1, KDSR |
| LW_B | 1 | 245 | 41940391 | 53513159 | MAN1A1, FAM184A, ASF1A, CEP85L, SLC35F1, NUS1, DCBLD1, ROS1, VGLL2, RFX6, GPRC6A, FAM162B, KPNA5, ZUP1, RSPH4A, PTP4A1, PHF3, ADGRB3, LMBRD1, COL19A1, COL9A1, FAM135A, SMAP1, B3GAT2, OGFRL1, RIMS1, KCNQ5, DPPA5, OOEP, CYB5R4, MRAP2, CEP162 |
| LW_B | 1 | 10 | 56563956 | 57069421 | RNGTT |
| LW_B | 1 | 11 | 61986232 | 62371709 | - |
| LW_B | 1 | 14 | 225017887 | 225614369 | CEMIP2, ABHD17B, C9orf85, GDA |
| LW_B | 1 | 18 | 227781084 | 229301982 | TRPM6, C9orf40, NMRK1, CARNMT1, OSTF1, PCSK5 |
| LW_B | 6 | 20 | 61261671 | 63135016 | PEG3, AURKC, ZNF304, ZNF772, ZNF773, ZNF550, ZNF606, ZNF135, ZNF329, ZNF274, ZNF8, RPS5, ZNF584, ZNF446, SLC27A5, ZBTB45, TRIM28, CHMP2A, UBE2M, MZF1 |
| LW_B | 6 | 84 | 107551091 | 113309905 | RBBP8, CABLES1, TMEM241, RIOK3, RMC1, NPC1, ANKRD29, LAMA3, TTC39C, CABYR, OSBPL1A, HRH4, ZNF521, SS18, PSMA8, TAF4B, KCTD1, AQP4, CHST9, CDH2 |
| LW_B | 10 | 14 | 28373981 | 28880540 | - |
| LW_B | 14 | 19 | 104827372 | 105533891 | MYOF, CEP55, FFAR4, RBP4, PDE6C, FRA10AC1, LGI1, SLC35G1, PLCE1 |
| LW_B | 15 | 22 | 26069754 | 26699994 | - |
| LW_A | LW_B | Gene | ||
|---|---|---|---|---|
| 1 | SSC1: 239389749–243367727 | 0.38 | 0.51 | NCBP1, PLPPR1, GRIN3A |
| 2 | SSC1: 155986286 | 0.73 | 0.31 | - |
| 3 | SSC4: 7921863 | 0.08 | 0.07 | - |
| 4 | SSC4:40772747–41434985 | 0.27 | 0.45 | - |
| 5 | SSC4: 119338511 | 0.23 | 0.31 | lncRNA |
| 6 | SSC5: 60515812–67077093 | 0.19 | 0.48 | ETV6, VWF, CCND2, TSPAN9 |
| 7 | SSC5: 10294138 | 0.12 | 0.31 | CARD10 |
| 8 | SSC6: 5154694 | 0.12 | 0.38 | CDH13 |
| 9 | SSC7: 103380850–115666493 | 0.24 | 0.07 | CEP128, SERPINA11 |
| 10 | SSC9: 106642564–119757434 | 0.08 | 0.52 | PIK3CG, COG5, BCAP29, SLC26A4 |
| 11 | SSC10: 49802418–50277330 | 0.23 | 0.31 | - |
| 12 | SSC11: 11335614 | 0.23 | 0.17 | NBEA |
| SSC11: 13345472–13527422 | 0.23 | 0.31 | TRPC4 | |
| SSC11: 65881573–69872868 | 0.30 | 0.66 | HS6ST3, NALCN | |
| 13 | SSC12: 48181049 | 0.12 | 0.38 | SMG6 |
| 14 | SSC13: 200778735–201245827 | 0.04 | 0.48 | TTC3, KCNJ6 |
| 15 | SSC14: 13891794 | 0.12 | 0.21 | - |
| 16 | SSC15: 115593713–121561503 | 0.23 | 0.38 | IKZF2, OBSL1 |
| 17 | SSC17: 8162613 | 0.19 | 0.31 | - |
| 18 | SSC18: 45801997 | 0.08 | 0.24 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolosov, A.; Getmantseva, L.; Kolosova, M.; Romanets, T.; Bakoev, N.; Romanets, E.; Bakoeva, I.; Kostyunina, O.; Prytkov, Y.; Tretiakova, O.; et al. Investigation of the Genetic Architecture of Pigs Subjected to Breeding Intensification. Genes 2022, 13, 197. https://doi.org/10.3390/genes13020197
Kolosov A, Getmantseva L, Kolosova M, Romanets T, Bakoev N, Romanets E, Bakoeva I, Kostyunina O, Prytkov Y, Tretiakova O, et al. Investigation of the Genetic Architecture of Pigs Subjected to Breeding Intensification. Genes. 2022; 13(2):197. https://doi.org/10.3390/genes13020197
Chicago/Turabian StyleKolosov, Anatoly, Lyubov Getmantseva, Maria Kolosova, Timofey Romanets, Nekruz Bakoev, Elena Romanets, Ilona Bakoeva, Olga Kostyunina, Yuri Prytkov, Olga Tretiakova, and et al. 2022. "Investigation of the Genetic Architecture of Pigs Subjected to Breeding Intensification" Genes 13, no. 2: 197. https://doi.org/10.3390/genes13020197
APA StyleKolosov, A., Getmantseva, L., Kolosova, M., Romanets, T., Bakoev, N., Romanets, E., Bakoeva, I., Kostyunina, O., Prytkov, Y., Tretiakova, O., & Bakoev, S. (2022). Investigation of the Genetic Architecture of Pigs Subjected to Breeding Intensification. Genes, 13(2), 197. https://doi.org/10.3390/genes13020197