
Smith-Magenis Syndrome—Clinical Review, Biological Background and Related Disorders

 , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Clinical Features
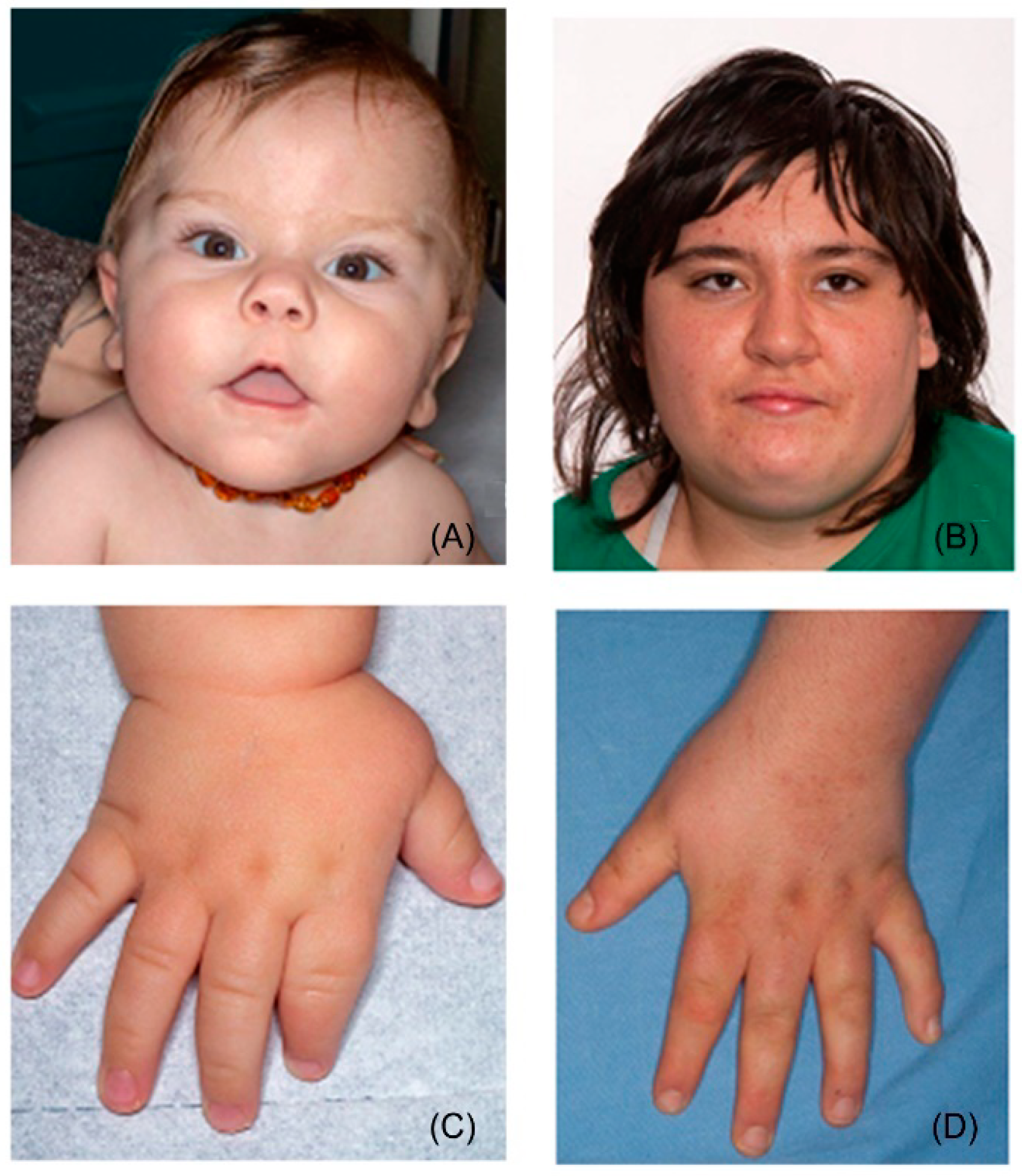
2.1. General Appearance
2.2. Growth
2.3. Multisystemic Manifestation
2.4. Neurodevelopmental Features
2.5. Behavioral Manifestations
2.6. Sleep Disorders
2.7. Neurological Problems
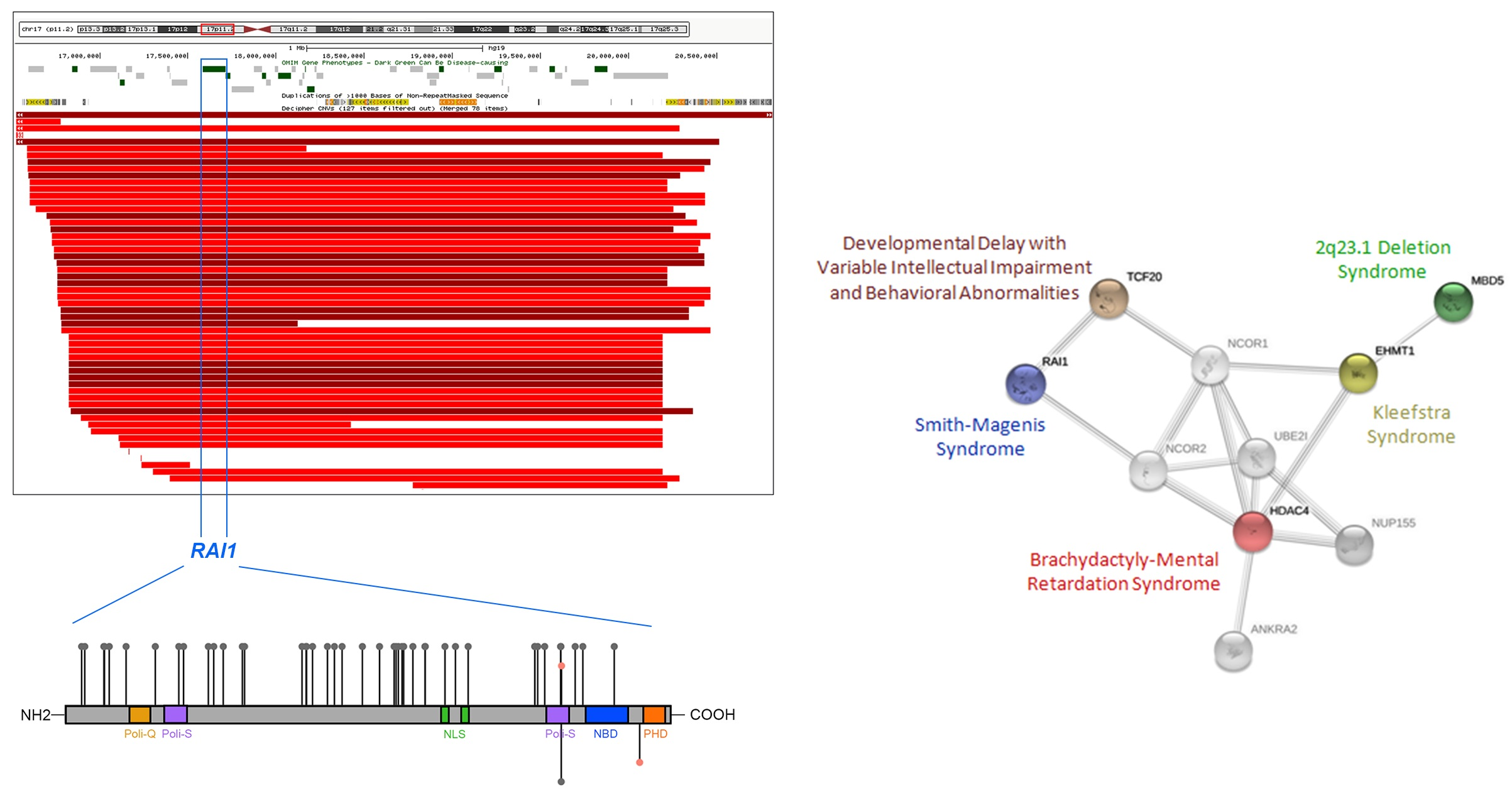
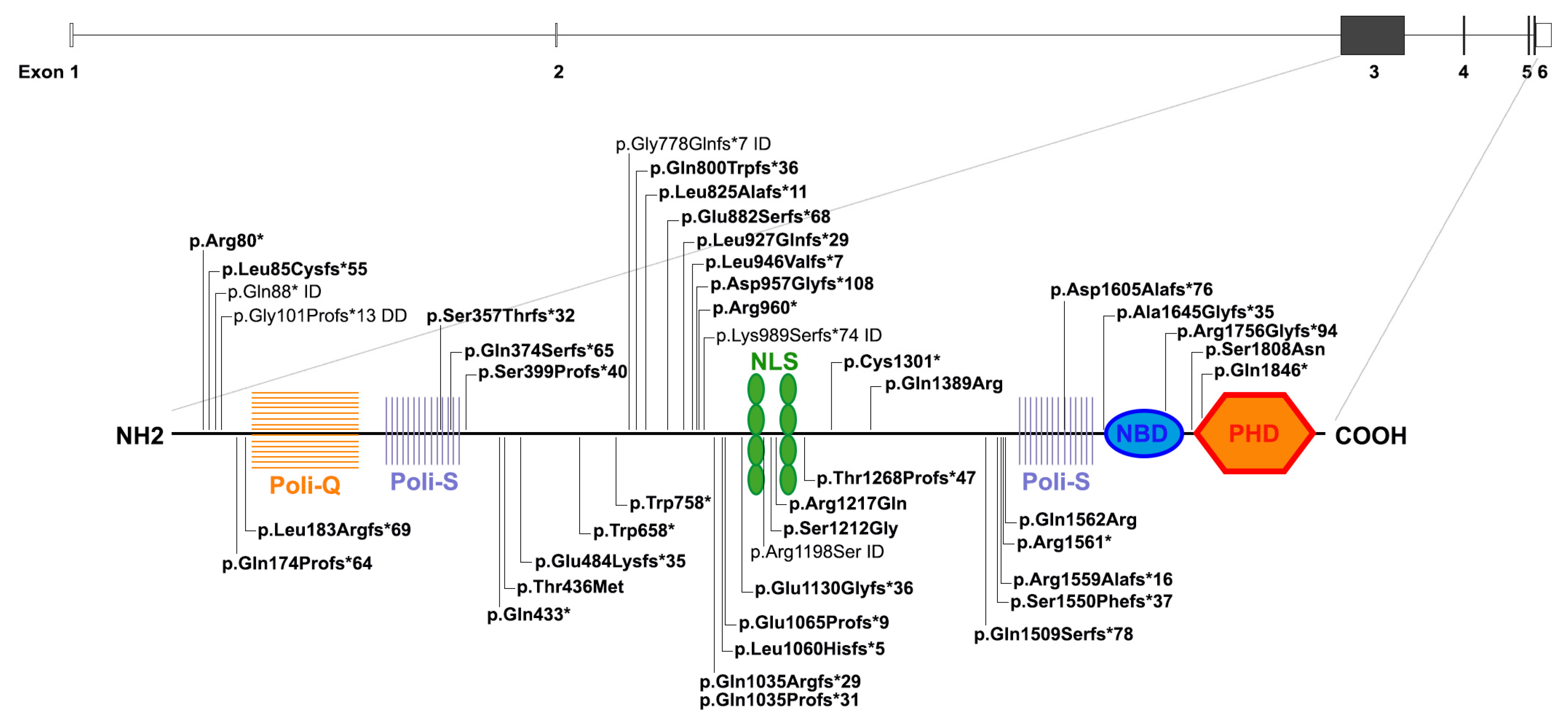
3. Genomic and Genetic Cause of SMS
4. Differential Diagnosis and Related Disorders
5. Treatment Insight
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allanson, J.E.; Greenberg, F.; Smith, A.C.M. The Face of Smith-Magenis Syndrome: A Subjective and Objective Study. J. Med. Genet. 1999, 36, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.M.; McGavran, L.; Robinson, J.; Waldstein, G.; Macfarlane, J.; Zonona, J.; Reiss, J.; Lahr, M.; Allen, L.; Magenis, E. Interstitial Deletion of (17)(p11.2p11.2) in Nine Patients. Am. J. Med. Genet. 1986, 24, 393–414. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, F.; Guzzetta, V.; De Oca-Luna, R.M.; Magenis, R.E.; Smith, A.C.M.; Richter, S.F.; Kondo, I.; Dobyns, W.B.; Patel, P.I.; Lupski, J.R. Molecular Analysis of the Smith-Magenis Syndrome: A Possible Contiguous-Gene Syndrome Associated with del(17)(p11.2). Am. J. Hum. Genet. 1991, 49, 1207–1218. [Google Scholar]
- Smith, A.C.M.; McGavran, L.; Waldstein, G. Deletion of the 17 Short Arm in Two Patients with Facial Clefts. Am. J. Med. Genet. 1982, 34, 410A. [Google Scholar]
- Slager, R.E.; Newton, T.L.; Vlangos, C.N.; Finucane, B.; Elsea, S.H. Mutations in RAI1 Associated with Smith-Magenis Syndrome. Nat. Genet. 2003, 33, 466–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.C.M.; Boyd, K.E.; Brennan, C.; Charles, J.; Elsea, S.H.; Finucane, B.M.; Foster, R.; Gropman, A.; Girirajan, S.; Haas-Givler, B. Smith-Magenis Syndrome. In GeneReviews® [Internet]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1310/ (accessed on 9 December 2021).
- Smith, A.C.M.; Magenis, R.E.; Elsea, S.H. Overview of Smith-Magenis Syndrome. J. Assoc. Genet. Technol. 2005, 31, 163–167. [Google Scholar] [PubMed]
- Edelman, E.A.; Girirajan, S.; Finucane, B.; Patel, P.I.; Lupski, J.R.; Smith, A.C.M.; Elsea, S.H. Gender, Genotype, and Phenotype Differences in Smith-Magenis Syndrome: A Meta-Analysis of 105 Cases. Clin. Genet. 2007, 71, 540–550. [Google Scholar] [CrossRef]
- Greenberg, F.; Lewis, R.A.; Potocki, L.; Glaze, D.; Parke, J.; Killian, J.; Murphy, M.A.; Williamson, D.; Brown, F.; Dutton, R.; et al. Multi-Disciplinary Clinical Study of Smith-Magenis Syndrome (Deletion 17p11.2). Am. J. Med. Genet. 1996, 62, 247–254. [Google Scholar] [CrossRef]
- Elsea, S.H.; Girirajan, S.S. Smith-Magenis Syndrome. Eur. J. Hum. Genet. 2008, 16, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Girirajan, S.; Elsas, L.J.; Devriendt, K.; Elsea, S.H. RAI1 Variations in Smith-Magenis Syndrome Patients without 17p11.2 Deletions. J. Med. Genet. 2005, 42, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Guérin-Moreau, M.; Colin, E.; Nguyen, S.; Andrieux, J.; De Leersnyder, H.; Bonneau, D.; Martin, L. Dermatologic Features of Smith-Magenis Syndrome. Pediatr. Dermatol. 2015, 32, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.M.; Lupski, J.R.; Greenberg, F.; Lewis, R.A. Ophthalmic Manifestations of Smith-Magenis Syndrome. Ophthalmology 1996, 103, 1084–1091. [Google Scholar] [CrossRef]
- Finucane, B.M.; Jaeger, E.R.; Kurtz, M.B.; Weinstein, M.; Scott, C.I. Eye Abnormalities in the Smith-Magenis Contiguous Gene Deletion Syndrome. Am. J. Med. Genet. 1993, 45, 443–446. [Google Scholar] [CrossRef]
- Gropman, A.L.; Duncan, W.C.; Smith, A.C.M. Neurologic and Developmental Features of the Smith-Magenis Syndrome (del 17p11.2). Pediatr. Neurol. 2006, 34, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.S.; Potocki, L.; Lupski, J.R. The Smith-Magenis Syndrome [del(17)p11.2]: Clinical Review and Molecular Advances. Ment. Retard. Dev. Disabil. Res. Rev. 1996, 2, 122–129. [Google Scholar] [CrossRef]
- Tomona, N.; Smith, A.C.M.; Guadagnini, J.P.; Hart, T.C. Craniofacial and Dental Phenotype of Smith-Magenis Syndrome. Am. J. Med. Genet. Part A 2006, 140, 2556–2561. [Google Scholar] [CrossRef] [PubMed]
- Di Cicco, M.; Padoan, R.; Felisati, G.; Dilani, D.; Moretti, E.; Guerneri, S.; Selicorni, A. Otorhinolaringologic Manifestation of Smith-Magenis Syndrome. Int. J. Pediatr. Otorhinolaryngol. 2001, 59, 147–150. [Google Scholar] [CrossRef]
- Smith, A.C.M.; Dykens, E.; Greenberg, F. Behavioral Phenotype of Smith-Magenis Syndrome (del 17p11.2). Am. J. Med. Genet. Neuropsychiatr. Genet. 1998, 81, 179–185. [Google Scholar] [CrossRef]
- La Guía, I.H.D.; Garayzábal-Heinze, E.; Gómez-Vilda, P. Voice Characteristics in Smith–Magenis Syndrome: An Acoustic Study of Laryngeal Biomechanics. Languages 2020, 5, 31. [Google Scholar] [CrossRef]
- Girirajan, S.; Vlangos, C.N.; Szomju, B.B.; Edelman, E.; Trevors, C.D.; Dupuis, L.; Nezarati, M.; Bunyan, D.J.; Elsea, S.H. Genotype-Phenotype Correlation in Smith-Magenis Syndrome: Evidence That Multiple Genes in 17p11.2 Contribute to the Clinical Spectrum. Genet. Med. 2006, 8, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Kondo, I.; Matsuura, S.; Kuwajima, K.; Tokashiki, M.; Izumikawa, Y.; Naritomi, K.; Niikawa, N.; Kajii, T. Diagnostic Hand Anomalies in Smith-Magenis Syndrome: Four New Patients with del (17)(p11.2p11.2). Am. J. Med. Genet. 1991, 41, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, A.E.; Potocki, L.; Poznanski, A.K.; Lupski, J.R. The Hand in Smith-Magenis Syndrome (Deletion 17p11.2): Evaluation by Metacarpophalangeal Pattern Profile Analysis. Pediatr. Radiol. 2003, 33, 173–176. [Google Scholar] [CrossRef] [PubMed]
- MarianneJensen, L.; Kirchhoff, M. Polydactyly in a Boy with Smith-Magenis Syndrome. Clin. Dysmorphol. 2005, 14, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Rive Le Gouard, N.; Jacquinet, A.; Ruaud, L.; Deleersnyder, H.; Ageorges, F.; Gallard, J.; Lacombe, D.; Odent, S.; Mikaty, M.; Manouvrier-Hanu, S.; et al. Smith-Magenis Syndrome: Clinical and Behavioral Characteristics in a Large Retrospective Cohort. Clin. Genet. 2021, 99, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Crain, C.A. An Assessment of Obesity and Hyperphagia in Individuals with Smith-Magenis Syndrome. Master’s Thesis, University of Texas Graduate School of Biomedical Sciences, Houston, TX, USA, May 2010. [Google Scholar]
- Burns, B.; Schmidt, K.; Williams, S.R.; Kim, S.; Girirajan, S.; Elsea, S.H. RAI1 Haploinsufficiency Causes Reduced Bdnf Expression Resulting in Hyperphagia, Obesity and Altered Fat Distribution in Mice and Humans with No Evidence of Metabolic Syndrome. Hum. Mol. Genet. 2010, 19, 4026–4042. [Google Scholar] [CrossRef] [Green Version]
- Potocki, L.; Shaw, C.J.; Stankiewicz, P.; Lupski, J.R. Variability in Clinical Phenotype despite Common Chromosomal Deletion in Smith-Magenis Syndrome [del(17)(p11.2p11.2)]. Genet. Med. 2003, 5, 430–434. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.C.M.; Gropman, A.L. Smith-Magenis Syndrome. In Cassidy and Allanson’s Management of Genetic Syndromes; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2021; pp. 863–893. [Google Scholar]
- De Leersnyder, H.; De Blois, M.C.; Claustrat, B.; Romana, S.; Albrecht, U.; Von Kleist-Retzow, J.C.; Delobel, B.; Viot, G.; Lyonnet, S.; Vekemans, M.; et al. Inversion of the Circadian Rhythm of Melatonin in the Smith-Magenis Syndrome. J. Pediatr. 2001, 139, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Hayashi, M.; Hasegawa, T.; Shimohira, M.; Kohyama, J. Systemic Growth Hormone Corrects Sleep Disturbance in Smith-Magenis Syndrome. Brain Dev. 2004, 26, 484–486. [Google Scholar] [CrossRef]
- Spadoni, E.; Colapietro, P.; Bozzola, M.; Marseglia, G.L.; Repossi, L.; Danesino, C.; Larizza, L.; Maraschio, P. Smith-Magenis Syndrome and Growth Hormone Deficiency. Eur. J. Pediatr. 2004, 163, 353–358. [Google Scholar] [CrossRef]
- Stratton, R.F.; Dobyns, W.B.; Greenberg, F.; DeSana, J.B.; Moore, C.; Fidone, G.; Runge, G.H.; Feldman, P.; Sekhon, G.S.; Pauli, R.M. Interstitial Deletion of (17)(p11.2p11.2): Report of Six Additional Patients with a New Chromosome Deletion Syndrome. Am. J. Med. Genet. 1986, 24, 421–432. [Google Scholar] [CrossRef]
- Onesimo, R.; Versacci, P.; Delogu, A.B.; De Rosa, G.; Pugnaloni, F.; Blandino, R.; Leoni, C.; Calcagni, G.; Digilio, M.C.; Zollino, M.; et al. Smith–Magenis Syndrome: Report of Morphological and New Functional Cardiac Findings with Review of the Literature. Am. J. Med. Genet. Part A 2021, 185, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Chou, I.C.; Tsai, F.J.; Yu, M.T.; Tsai, C.H. Smith-Magenis Syndrome with Bilateral Vesicoureteral Reflux: A Case Report. J. Formos. Med. Assoc. 2002, 101, 726–728. [Google Scholar] [PubMed]
- Fischer, H.; Oswald, H.-P.; Duba, H.-C.; Doczy, L.; Simma, B.; Utermann, G.; Haas, O. Constitutional Interstitial Deletion of 17(p11.2) (Smith-Magenis Syndrome): A Clinically Recognizable Microdeletion Syndrome. Klin. Pädiatr. 1993, 205, 166–170. [Google Scholar] [CrossRef]
- Al-Qudah, A.A.; El-Khateeb, M.S.; Abu-Hamour, W.; Bulos, N.K. Smith-Magenis Syndrome: Report of Two Cases and Review of the Literature. Ann. Saudi Med. 1994, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.M.; Challman, T.D.; Bock, G.H. End-Stage Renal Failure in Smith-Magenis Syndrome. Am. J. Med. Genet. Part A 2007, 143, 1922–1924. [Google Scholar] [CrossRef]
- Fan, Y.-S.; Farrell, S.A. Prenatal Diagnosis of Interstitial Deletion of 17(p11.2p11.2) (Smith-Magenis Syndrome). Am. J. Med. Genet. 1994, 49, 253–254. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Tian, H.; Rong, L.; Wang, F.; Yu, X.; Li, Y.; Gao, J. Prenatal Diagnosis and Neonatal Phenotype of a de novo Microdeletion of 17p11.2p12 Associated with Smith-Magenis Syndrome and External Genital Defects. J. Genet. 2020, 99, 50. [Google Scholar] [CrossRef]
- Brendal, M.A.; King, K.A.; Zalewski, C.K.; Finucane, B.M.; Introne, W.; Brewer, C.C.; Smith, A.C.M. Auditory Phenotype of Smith–Magenis Syndrome. J. Speech Lang. Hear. Res. 2017, 60, 1076–1087. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo de la Guía, I.; Garayzábal Heinze, E. Diferencias Fonológicas Entre Síndromes Del Neurodesarrollo: Evidencias a Partir de Los Procesos de Simplificación Fonológica Más Frecuentes. Rev. Investig. Logop. 2019, 9, 81–106. [Google Scholar] [CrossRef]
- Spilsbury, J.; Mohanty, K. The Orthopaedic Manifestations of Smith-Magenis Syndrome. J. Pediatr. Orthop. B 2003, 12, 22–26. [Google Scholar] [CrossRef]
- Perkins, T.; Rosenberg, J.M.; Coz, C.L.; Alaimo, J.T.; Trofa, M. Smith-Magenis Syndrome Patients Often Display Antibody Deficiency but Not Other Immune Pathologies. J. Allergy Clin. Immunol. Pract. 2017, 5, 1344–1350.e3. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.M.; Groptnan, A.L.; Bailey-Wilson, J.E.; Goker-Alpan, O.; Elsea, S.H.; Blancato, J.; Lupski, J.R.; Potocki, L. Hypercholesterolemia in Children with Smith-Magenis Syndrome: Del (17)(p11.2p11.2). Genet. Med. 2002, 4, 118–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, L.S.; Linehan, W.M. Molecular Genetics and Clinical Features of Birt-Hogg-Dubé Syndrome. Nat. Rev. Urol. 2015, 12, 558–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, H.T.; Dudding, T.; Blanchard, C.L.; Elsea, S.H. Frameshift Mutation Hotspot Identified in Smith-Magenis Syndrome: Case Report and Review of Literature. BMC Med. Genet. 2010, 11, 142. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.C.M.; Fleming, L.R.; Piskorski, A.M.; Amin, A.; Phorphutkul, C.; de la Monte, S.; Stopa, E.; Introne, W.; Vilboux, T.; Duncan, F.; et al. Deletion of 17p11.2 Encompasses FLCN with Increased Risk of Birt-Hogg-Dubé in Smith Magenis Syndrome: Recommendation for Cancer Screening. Abstract 2603S. In Proceedings of the 63rd Annual Meeting of The American Society of Human Genetics, San Diego, CA, USA, 18–22 October 2014. [Google Scholar]
- Dardour, L.; Verleyen, P.; Lesage, K.; Holvoet, M.; Devriendt, K. Bilateral Renal Tumors in an Adult Man with Smith-Magenis Syndrome: The Role of the FLCN Gene. Eur. J. Med. Genet. 2016, 59, 499–501. [Google Scholar] [CrossRef]
- Finucane, B.; Savatt, J.M.; Shimelis, H.; Girirajan, S.; Myers, S.M. Birt-Hogg-Dubé Symptoms in Smith-Magenis Syndrome Include Pediatric-Onset Pneumothorax. Am. J. Med. Genet. Part A 2021, 185, 1922–1924. [Google Scholar] [CrossRef]
- Wolters, P.L.; Gropman, A.L.; Martin, S.C.; Smith, M.R.; Hildenbrand, H.L.; Brewer, C.C.; Smith, A.C.M. Neurodevelopment of Children Under 3 Years of Age with Smith-Magenis Syndrome. Pediatr. Neurol. 2009, 41, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Madduri, N.; Peters, S.U.; Voigt, R.G.; Llorente, A.M.; Lupski, J.R.; Potocki, L. Cognitive and Adaptive Behavior Profiles in Smith-Magenis Syndrome. J. Dev. Behav. Pediatr. 2006, 27, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Udwin, O.; Webber, C.; Horn, I. Abilities and Attainment in Smith-Magenis Syndrome. Dev. Med. Child Neurol. 2001, 43, 823. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.C.; Wolters, P.L.; Smith, A.C.M. Adaptive and Maladaptive Behavior in Children with Smith-Magenis Syndrome. J. Autism Dev. Disord. 2006, 36, 541–552. [Google Scholar] [CrossRef]
- Garayzábal Heinze, E.; Lens Villaverde, M.; Moruno López, E.; Conde Magro, T.; Moura, L.F.; Fernández, M.; Sampaio, A. General cognitive functioning and psycholinguistic abilities in children with Smith-Magenis Syndrome. Psicothema 2011, 23, 725–731. [Google Scholar] [PubMed]
- Dykens, E.M.; Finucane, B.M.; Gayley, C. Brief Report: Cognitive and Behavioral Profiles in Persons with Smith-Magenis Syndrome. J. Autism Dev. Disord. 1997, 27, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Osório, A.; Cruz, R.; Sampaio, A.; Garayzábal, E.; Carracedo, Á.; Fernández-Prieto, M. Cognitive Functioning in Children and Adults with Smith-Magenis Syndrome. Eur. J. Med. Genet. 2012, 55, 394–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poisson, A.; Nicolas, A.; Cochat, P.; Sanlaville, D.; Rigard, C.; De Leersnyder, H.; Franco, P.; Portes, V.D.; Edery, P.; Demily, C. Behavioral Disturbance and Treatment Strategies in Smith-Magenis Syndrome. Orphanet J. Rare Dis. 2015, 10, 111. [Google Scholar] [CrossRef] [Green Version]
- Laje, G.; Morse, R.; Richter, W.; Ball, J.; Pao, M.; Smith, A.C.M. Autism Spectrum Features in Smith-Magenis Syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154, 456–462. [Google Scholar] [CrossRef]
- Nag, H.E.; Nordgren, A.; Anderlid, B.-M.; Nærland, T. Reversed Gender Ratio of Autism Spectrum Disorder in Smith-Magenis Syndrome. Mol. Autism 2018, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Bissell, S.; Wilde, L.; Richards, C.; Moss, J.; Oliver, C. The Behavioural Phenotype of Potocki-Lupski Syndrome: A Cross-Syndrome Comparison. J. Neurodev. Disord. 2018, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Hildenbrand, H.L.; Smith, A.C.M. Analysis of the Sensory Profile in Children with Smith-Magenis Syndrome. Phys. Occup. Ther. Pediatr. 2012, 32, 48–65. [Google Scholar] [CrossRef]
- Dykens, E.M.; Smith, A.C.M. Distinctiveness and Correlates of Maladaptive Behaviour in Children and Adolescents with Smith-Magenis Syndrome. J. Intellect. Disabil. Res. 1998, 42, 481–489. [Google Scholar] [CrossRef]
- Finucane, B.; Dirrigl, K.H.; Simon, E.W. Characterization of Self-Injurious Behaviors in Children and Adults with Smith-Magenis Syndrome. Am. J. Ment. Retard. 2001, 106, 52–58. [Google Scholar] [CrossRef]
- Nag, H.E.; Nærland, T. Age-Related Changes in Behavioural and Emotional Problems in Smith–Magenis Syndrome Measured with the Developmental Behavior Checklist. J. Intellect. Disabil. 2020, 25, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, L.; Oliver, C. The Behavioural Phenotype of Smith-Magenis Syndrome: Evidence for a Gene-Environment Interaction. J. Intellect. Disabil. Res. 2008, 52, 830–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finucane, B.; Haas-Givler, B. Smith-Magenis Syndrome: Genetic Basis and Clinical Implications. J. Ment. Health Res. Intellect. Disabil. 2009, 2, 134–148. [Google Scholar] [CrossRef]
- Shayota, B.J.; Elsea, S.H. Behavior and Sleep Disturbance in Smith-Magenis Syndrome. Curr. Opin. Psychiatry 2019, 32, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Sloneem, J.; Oliver, C.; Udwin, O.; Woodcock, K.A. Prevalence, Phenomenology, Aetiology and Predictors of Challenging Behaviour in Smith-Magenis Syndrome. J. Intellect. Disabil. Res. 2011, 55, 138–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaimo, J.T.; Barton, L.V.; Mullegama, S.V.; Wills, R.D.; Foster, R.H.; Elsea, S.H. Individuals with Smith-Magenis Syndrome Display Profound Neurodevelopmental Behavioral Deficiencies and Exhibit Food-Related Behaviors Equivalent to Prader-Willi Syndrome. Res. Dev. Disabil. 2015, 47, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Finucane, B.M.; Konar, D.; Givler, B.H.; Kurtt, M.B.; Scott, C.I. The Spasmodic Upper-body Squeeze: A Chalacteristic Behavior in Smith-Magenis Syndrome. Dev. Med. Child Neurol. 1994, 36, 78–83. [Google Scholar] [CrossRef]
- Greenberg, F.; Magenis, W.; Finucane, B.M.; Smith, A.C.M.; Patel, P.I.; Lupski, J.R. Smith-Magenis Syndrome [del(17)(p11.2)] and Its Clinical Overlap with Prader-Willi Syndrome. Abstract. In Proceedings of the 45th Annual Meeting of The American Society of Human Genetics, San Francisco, CA, USA, 24–28 October 1996. [Google Scholar]
- Smith, A.C.M.; Dykens, E.; Greenberg, F. Sleep Disturbance in Smith-Magenis Syndrome (del 17 p11.2). Am. J. Med. Genet. (Neuropsychiatr. Genet.) 1998, 81, 186–191. [Google Scholar] [CrossRef]
- Smith, A.C.M.; Morse, R.S.; Introne, W.; Duncan, W.C. Twenty-Four-Hour Motor Activity and Body Temperature Patterns Suggest Altered Central Circadian Timekeeping in Smith–Magenis Syndrome, a Neurodevelopmental Disorder. Am. J. Med. Genet. Part A 2019, 179, 224–236. [Google Scholar] [CrossRef]
- Agar, G.; Brown, C.; Sutherland, D.; Coulborn, S.; Oliver, C.; Richards, C. Sleep Disorders in Rare Genetic Syndromes: A Meta-Analysis of Prevalence and Profile. Mol. Autism 2021, 12, 18. [Google Scholar] [CrossRef]
- Boudreau, E.A.; Johnson, K.P.; Jackman, A.R.; Blancato, J.; Huizing, M.; Bendavid, C.; Jones, M.P.; Chandrasekharappa, S.C.; Lewy, A.J.; Smith, A.C.M.; et al. Review of Disrupted Sleep Patterns in Smith-Magenis Syndrome and Normal Melatonin Secretion in a Patient with an Atypical Interstitial 17p11.2 Deletion. Am. J. Med. Genet. Part A 2009, 149, 1382–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potocki, L.; Glaze, D.; Tan, D.X.; Park, S.S.; Kashork, C.D.; Shaffer, L.G.; Reiter, R.J.; Lupski, J.R. Circadian Rhythm Abnormalities of Melatonin in Smith-Magenis Syndrome. J. Med. Genet. 2000, 37, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.M.; Potocki, L.; Walz, K.; Lynch, J.K.; Glaze, D.G.; Lupski, J.R.; Noebels, J.L. Epilepsy and Chromosomal Rearrangements in Smith-Magenis Syndrome [del(17)(p11.2p11.2)]. J. Child Neurol. 2006, 21, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Boddaert, N.; De Leersnyder, H.; Bourgeois, M.; Munnich, A.; Brunelle, F.; Zilbovicius, M. Anatomical and Functional Brain Imaging Evidence of Lenticulo-Insular Anomalies in Smith Magenis Syndrome. NeuroImage 2004, 21, 1021–1025. [Google Scholar] [CrossRef]
- Masuno, M.; Asano, J.; Arai, M.; Kuwahara, T.; Orii, T. Interstitial Deletion of 17p11.2 with Brain Abnormalities. Clin. Genet. 2008, 41, 278–280. [Google Scholar] [CrossRef]
- Natacci, F.; Corrado, L.; Pierri, M.; Rossetti, M.; Zuccarini, C.; Riva, P.; Miozzo, M.; Larizza, L. Patient with Large 17p11.2 Deletion Presenting with Smith-Magenis Syndrome and Joubert Syndrome Phenotype. Am. J. Med. Genet. 2000, 95, 467–472. [Google Scholar] [CrossRef]
- Capra, V.; Biancheri, R.; Morana, G.; Striano, P.; Novara, F.; Ferrero, G.B.; Boeri, L.; Celle, M.E.; Mancardi, M.M.; Zuffardi, O.; et al. Periventricular Nodular Heterotopia in Smith-Magenis Syndrome. Am. J. Med. Genet. Part A 2014, 164, 3142–3147. [Google Scholar] [CrossRef]
- Girirajan, S.; Mendoza-Londono, R.; Vlangos, C.N.; Dupuis, L.; Nowak, N.J.; Bunyan, D.J.; Hatchwell, E.; Elsea, S.H. Smith-Magenis Syndrome and Moyamoya Disease in a Patient with del(17)(p11.2p13.1). Am. J. Med. Genet. Part A 2007, 143, 999–1008. [Google Scholar] [CrossRef]
- Chaudhry, A.P.; Schwartz, C.; Singh, A.K. Stroke after Cardiac Surgery in a Patient with Smith-Magenis Syndrome. Tex. Heart Inst. J. 2007, 34, 247–249. [Google Scholar]
- Vlangos, C. Refinement of the Smith–Magenis Syndrome Critical Region to ∼950 kb and Assessment of 17p11.2 Deletions. Are All Deletions Created Equally? Mol. Genet. Metab. 2003, 79, 134–141. [Google Scholar] [CrossRef]
- Elsea, S.H.; Williams, S.R. Smith-Magenis Syndrome: Haploinsufficiency of RAI1 Results in Altered Gene Regulation in Neurological and Metabolic Pathways. Expert Rev. Mol. Med. 2011, 13, e14. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; Stankiewicz, P. Genomic Disorders: Molecular Mechanisms for Rearrangements and Conveyed Phenotypes. PLoS Genet. 2005, 1, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boot, E.; Linders, C.C.; Tromp, S.H.; van den Boogaard, M.J.; van Eeghen, A.M. Possible Underreporting of Pathogenic Variants in RAI1 Causing Smith–Magenis Syndrome. Am. J. Med. Genet. Part A 2021, 185, 3167–3169. [Google Scholar] [CrossRef] [PubMed]
- Falco, M.; Amabile, S.; Acquaviva, F. RAI1 Gene Mutations: Mechanisms of Smith-Magenis Syndrome. Appl. Clin. Genet. 2017, 10, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Zori, R.T.; Lupski, J.R.; Heju, Z.; Greenberg, F.; Killian, J.M.; Gray, B.A.; Driscoll, D.J.; Patel, P.I.; Zackowski, J.L. Clinical, Cytogenetic, and Molecular Evidence for an Infant with Smith–Magenis Syndrome Born from a Mother Having a Mosaic 17p11.2p12 Deletion. Am. J. Med. Genet. 1993, 47, 504–511. [Google Scholar] [CrossRef]
- Yang, S.P.; Bidichandani, S.I.; Figuera, L.E.; Juyal, R.C.; Saxon, P.J.; Baldini, A.; Patel, P.I. Molecular Analysis of Deletion (17)(p11.2p11.2) in a Family Segregating a 17p Paracentric Inversion: Implications for Carriers of Paracentric Inversions. Am. J. Hum. Genet. 1997, 60, 1184–1193. [Google Scholar]
- Campbell, I.M.; Yuan, B.; Robberecht, C.; Pfundt, R.; Szafranski, P.; McEntagart, M.E.; Nagamani, S.C.S.; Erez, A.; Bartnik, M.; Wiśniowiecka-Kowalnik, B.; et al. Parental Somatic Mosaicism Is Underrecognized and Influences Recurrence Risk of Genomic Disorders. Am. J. Hum. Genet. 2014, 95, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Park, J.P.; Moeschler, J.B.; Davies, W.S.; Patel, P.I.; Mohandas, T.K. Smith-Magenis Syndrome Resulting from a de Novo Direct Insertion of Proximal 17q into 17p11.2. Am. J. Med. Genet. 1998, 77, 23–27. [Google Scholar] [CrossRef]
- Goh, E.S.-Y.; Banwell, B.; Stavropoulos, D.J.; Shago, M.; Yoon, G. Mosaic Microdeletion of 17p11.2-P12 and Duplication of 17q22-Q24 in a Girl with Smith-Magenis Phenotype and Peripheral Neuropathy. Am. J. Med. Genet. A 2014, 164, 748–752. [Google Scholar] [CrossRef]
- Acquaviva, F.; Sana, M.E.; Della Monica, M.; Pinelli, M.; Postorivo, D.; Fontana, P.; Falco, M.T.; Nardone, A.M.; Lonardo, F.; Iascone, M.; et al. First Evidence of Smith-Magenis Syndrome in Mother and Daughter Due to a Novel RAI Mutation. Am. J. Med. Genet. A 2017, 173, 231–238. [Google Scholar] [CrossRef]
- Carmona-Mora, P.; Walz, K. Retinoic Acid Induced 1, RAI1: A Dosage Sensitive Gene Related to Neurobehavioral Alterations Including Autistic Behavior. Curr. Genom. 2010, 11, 607–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Mora, P.; Encina, C.A.; Canales, C.P.; Cao, L.; Molina, J.; Kairath, P.; Young, J.I.; Walz, K. Functional and Cellular Characterization of Human Retinoic Acid Induced 1 (RAI1) Mutations Associated with Smith-Magenis Syndrome. BMC Mol. Biol 2010, 11, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvekar, S.; Johnsen, S.S.; Eriksen, A.B.; Johansen, T.; Sjøttem, E. Identification of Two Independent Nucleosome-Binding Domains in the Transcriptional Co-Activator SPBP. Biochem. J. 2012, 442, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvekar, S.; Rekdal, C.; Johansen, T.; Sjøttem, E. A Phylogenetic Study of SPBP and RAI1: Evolutionary Conservation of Chromatin Binding Modules. PLoS ONE 2013, 8, e78907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.-H.; Guenthner, C.J.; Xu, J.; Nguyen, T.; Schwarz, L.A.; Wilkinson, A.W.; Gozani, O.; Chang, H.Y.; Shamloo, M.; Luo, L. Molecular and Neural Functions of Rai1, the Causal Gene for Smith-Magenis Syndrome. Neuron 2016, 92, 392–406. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL Targets SET Domain Methyltransferase Activity to Hox Gene Promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Nakamura, T.; Mori, T.; Tada, S.; Krajewski, W.; Rozovskaia, T.; Wassell, R.; Dubois, G.; Mazo, A.; Croce, C.M.; Canaani, E. ALL-1 Is a Histone Methyltransferase That Assembles a Supercomplex of Proteins Involved in Transcriptional Regulation. Mol. Cell 2002, 10, 1119–1128. [Google Scholar] [CrossRef]
- Bi, W.; Saifi, G.M.; Shaw, C.J.; Walz, K.; Fonseca, P.; Wilson, M.; Potocki, L.; Lupski, J.R. Mutations of RAI1, a PHD-Containing Protein, in Nondeletion Patients with Smith-Magenis Syndrome. Hum. Genet. 2004, 115, 515–524. [Google Scholar] [CrossRef]
- Garay, P.M.; Chen, A.; Tsukahara, T.; Rodríguez Díaz, J.C.; Kohen, R.; Althaus, J.C.; Wallner, M.A.; Giger, R.J.; Jones, K.S.; Sutton, M.A.; et al. RAI1 Regulates Activity-Dependent Nascent Transcription and Synaptic Scaling. Cell Rep. 2020, 32, 108002. [Google Scholar] [CrossRef]
- Williams, S.R.; Zies, D.; Mullegama, S.V.; Grotewiel, M.S.; Elsea, S.H. Smith-Magenis Syndrome Results in Disruption of CLOCK Gene Transcription and Reveals an Integral Role for RAI1 in the Maintenance of Circadian Rhythmicity. Am. J. Hum. Genet. 2012, 90, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Garay, P.M.; Wallner, M.A.; Iwase, S. Yin–Yang Actions of Histone Methylation Regulatory Complexes in the Brain. Epigenomics 2016, 8, 1689–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberl, H.C.; Spruijt, C.G.; Kelstrup, C.D.; Vermeulen, M.; Mann, M. A Map of General and Specialized Chromatin Readers in Mouse Tissues Generated by Label-Free Interaction Proteomics. Mol. Cell 2013, 49, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, W.D.; Dafou, D.; McEntagart, M.; Woollard, W.J.; Elmslie, F.V.; Holder-Espinasse, M.; Irving, M.; Saggar, A.K.; Smithson, S.; Trembath, R.C.; et al. De Novo Mutations in MLL Cause Wiedemann-Steiner Syndrome. Am. J. Hum. Genet. 2012, 91, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Babbs, C.; Lloyd, D.; Pagnamenta, A.T.; Twigg, S.R.F.; Green, J.; McGowan, S.J.; Mirza, G.; Naples, R.; Sharma, V.P.; Volpi, E.V.; et al. De Novo and Rare Inherited Mutations Implicate the Transcriptional Coregulator TCF20/SPBP in Autism Spectrum Disorder. J. Med. Genet. 2014, 51, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.R.; Aldred, M.A.; Der Kaloustian, V.M.; Halal, F.; Gowans, G.; McLeod, D.R.; Zondag, S.; Toriello, H.V.; Magenis, R.E.; Elsea, S.H. Haploinsufficiency of HDAC4 Causes Brachydactyly Mental Retardation Syndrome, with Brachydactyly Type E, Developmental Delays, and Behavioral Problems. Am. J. Hum. Genet. 2010, 87, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.R.; Mullegama, S.V.; Rosenfeld, J.A.; Dagli, A.I.; Hatchwell, E.; Allen, W.P.; Williams, C.A.; Elsea, S.H. Haploinsufficiency of MBD5 Associated with a Syndrome Involving Microcephaly, Intellectual Disabilities, Severe Speech Impairment, and Seizures. Eur. J. Hum. Genet. 2010, 18, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talkowski, M.E.; Mullegama, S.V.; Rosenfeld, J.A.; van Bon, B.W.M.; Shen, Y.; Repnikova, E.A.; Gastier-Foster, J.; Thrush, D.L.; Kathiresan, S.; Ruderfer, D.M.; et al. Assessment of 2q23.1 Microdeletion Syndrome Implicates MBD5 as a Single Causal Locus of Intellectual Disability, Epilepsy, and Autism Spectrum Disorder. Am. J. Hum. Genet. 2011, 89, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Mullegama, S.V.; Alaimo, J.T.; Chen, L.; Elsea, S.H. Phenotypic and Molecular Convergence of 2q23.1 Deletion Syndrome with Other Neurodevelopmental Syndromes Associated with Autism Spectrum Disorder. Int. J. Mol. Sci. 2015, 16, 7627–7643. [Google Scholar] [CrossRef] [Green Version]
- Mullegama, S.V.; Pugliesi, L.; Burns, B.; Shah, Z.; Tahir, R.; Gu, Y.; Nelson, D.L.; Elsea, S.H. MBD5 Haploinsufficiency Is Associated with Sleep Disturbance and Disrupts Circadian Pathways Common to Smith-Magenis and Fragile X Syndromes. Eur. J. Hum. Genet. 2015, 23, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Kleefstra, T.; Kramer, J.M.; Neveling, K.; Willemsen, M.H.; Koemans, T.S.; Vissers, L.E.L.M.; Wissink-Lindhout, W.; Fenckova, M.; van den Akker, W.M.R.; Kasri, N.N.; et al. Disruption of an EHMT1-Associated Chromatin-Modification Module Causes Intellectual Disability. Am. J. Hum. Genet. 2012, 91, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Koemans, T.S.; Kleefstra, T.; Chubak, M.C.; Stone, M.H.; Reijnders, M.R.F.; de Munnik, S.; Willemsen, M.H.; Fenckova, M.; Stumpel, C.T.R.M.; Bok, L.A.; et al. Functional Convergence of Histone Methyltransferases EHMT1 and KMT2C Involved in Intellectual Disability and Autism Spectrum Disorder. PLoS Genet. 2017, 13, e1006864. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Brunner, H.G.; Amiel, J.; Oudakker, A.R.; Nillesen, W.M.; Magee, A.; Geneviève, D.; Cormier-Daire, V.; van Esch, H.; Fryns, J.-P.; et al. Loss-of-Function Mutations in Euchromatin Histone Methyl Transferase 1 (EHMT1) Cause the 9q34 Subtelomeric Deletion Syndrome. Am. J. Hum. Genet. 2006, 79, 370–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benevento, M.; Iacono, G.; Selten, M.; Ba, W.; Oudakker, A.; Frega, M.; Keller, J.; Mancini, R.; Lewerissa, E.; Kleefstra, T.; et al. Histone Methylation by the Kleefstra Syndrome Protein EHMT1 Mediates Homeostatic Synaptic Scaling. Neuron 2016, 91, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacono, G.; Dubos, A.; Méziane, H.; Benevento, M.; Habibi, E.; Mandoli, A.; Riet, F.; Selloum, M.; Feil, R.; Zhou, H.; et al. Increased H3K9 Methylation and Impaired Expression of Protocadherins Are Associated with the Cognitive Dysfunctions of the Kleefstra Syndrome. Nucleic Acids Res. 2018, 46, 4950–4965. [Google Scholar] [CrossRef]
- Loviglio, M.N.; Beck, C.R.; White, J.J.; Leleu, M.; Harel, T.; Guex, N.; Niknejad, A.; Bi, W.; Chen, E.S.; Crespo, I.; et al. Identification of a RAI1-Associated Disease Network through Integration of Exome Sequencing, Transcriptomics, and 3D Genomics. Genome Med. 2016, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.I.; Ciccone, C.; Simon, K.L.; Malicdan, M.C.; Vilboux, T.; Billington, C.; Fischer, R.; Introne, W.J.; Gropman, A.; Blancato, J.K.; et al. Exome Analysis of Smith–Magenis-like Syndrome Cohort Identifies de novo Likely Pathogenic Variants. Hum. Genet. 2017, 136, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Vetrini, F.; McKee, S.; Rosenfeld, J.A.; Suri, M.; Lewis, A.M.; Nugent, K.M.; Roeder, E.; Littlejohn, R.O.; Holder, S.; Zhu, W.; et al. De Novo and Inherited TCF20 Pathogenic Variants Are Associated with Intellectual Disability, Dysmorphic Features, Hypotonia, and Neurological Impairments with Similarities to Smith-Magenis Syndrome. Genome Med. 2019, 11, 12. [Google Scholar] [CrossRef]
- Kerkhof, J.; Squeo, G.M.; McConkey, H.; Levy, M.A.; Piemontese, M.R.; Castori, M.; Accadia, M.; Biamino, E.; Della Monica, M.; Di Giacomo, M.C.; et al. DNA Methylation Episignature Testing Improves Molecular Diagnosis of Mendelian Chromatinopathies. Genet. Med. 2022, 24, 51–60. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.-C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; Dubourg, C.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef]
- Laje, G.; Bernert, R.; Morse, R.; Pao, M.; Smith, A.C.M. Pharmacological Treatment of Disruptive Behavior in Smith-Magenis Syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.R.; Zinkstok, J.R.; Rommelse, N.N.J.; van de Ven, P.M.; Roes, K.C.B.; Wijburg, F.A.; de Rooij-Askes, E.; Linders, C.; Boot, E.; van Eeghen, A.M. Methylphenidate for Attention-Deficit/Hyperactivity Disorder in Patients with Smith–Magenis Syndrome: Protocol for a Series of N-of-1 Trials. Orphanet J. Rare Dis. 2021, 16, 380. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J. Psychopharmacological Interventions in Fragile X Syndrome, Fetal Alcohol Syndrome, Prader-Willi Syndrome, Angelman Syndrome, Smith-Magenis Syndrome, and Velocardiofacial Syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 1999, 5, 305–313. [Google Scholar] [CrossRef]
- Niederhofer, H. Efficacy of Risperidone Treatment in Smith-Magenis Syndrome (del 17 pll. 2). Psychiatr. Danub. 2007, 19, 189–192. [Google Scholar] [PubMed]
- Kaplan, K.A.; Elsea, S.H.; Potocki, L. Management of Sleep Disturbances Associated with Smith-Magenis Syndrome. CNS Drugs 2020, 34, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Williams Buckley, A.; Hirtz, D.; Oskoui, M.; Armstrong, M.J.; Batra, A.; Bridgemohan, C.; Coury, D.; Dawson, G.; Donley, D.; Findling, R.L.; et al. Practice Guideline: Treatment for Insomnia and Disrupted Sleep Behavior in Children and Adolescents with Autism Spectrum Disorder. Neurology 2020, 94, 392–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Leersnyder, H.; Bresson, J.L.; de Blois, M.-C.; Souberbielle, J.-C.; Mogenet, A.; Delhotal-Landes, B.; Salefranque, F.; Munnich, A. Beta 1-Adrenergic Antagonists and Melatonin Reset the Clock and Restore Sleep in a Circadian Disorder, Smith-Magenis Syndrome. J. Med. Genet. 2003, 40, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, C.M.; Brooks, J.; Czeisler, E.L.; Fisher, M.A.; Gibson, M.M.; Kite, K.; Smieszek, S.P.; Xiao, C.; Elsea, S.H.; Birznieks, G.; et al. Tasimelteon Safely and Effectively Improves Sleep in Smith-Magenis Syndrome: A Double-Blind Randomized Trial Followed by an Open-Label Extension. Genet. Med. 2021, 23, 2426–2432. [Google Scholar] [CrossRef]
- Richardson, G.S.; Zee, P.C.; Wang-Weigand, S.; Rodriguez, L.; Peng, X. Circadian Phase-Shifting Effects of Repeated Ramelteon Administration in Healthy Adults. J. Clin. Sleep Med. 2008, 4, 456–461. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinaldi, B.; Villa, R.; Sironi, A.; Garavelli, L.; Finelli, P.; Bedeschi, M.F. Smith-Magenis Syndrome—Clinical Review, Biological Background and Related Disorders. Genes 2022, 13, 335. https://doi.org/10.3390/genes13020335
Rinaldi B, Villa R, Sironi A, Garavelli L, Finelli P, Bedeschi MF. Smith-Magenis Syndrome—Clinical Review, Biological Background and Related Disorders. Genes. 2022; 13(2):335. https://doi.org/10.3390/genes13020335
Chicago/Turabian StyleRinaldi, Berardo, Roberta Villa, Alessandra Sironi, Livia Garavelli, Palma Finelli, and Maria Francesca Bedeschi. 2022. "Smith-Magenis Syndrome—Clinical Review, Biological Background and Related Disorders" Genes 13, no. 2: 335. https://doi.org/10.3390/genes13020335
APA StyleRinaldi, B., Villa, R., Sironi, A., Garavelli, L., Finelli, P., & Bedeschi, M. F. (2022). Smith-Magenis Syndrome—Clinical Review, Biological Background and Related Disorders. Genes, 13(2), 335. https://doi.org/10.3390/genes13020335