
Leveraging Gene-Level Prediction as Informative Covariate in Hypothesis Weighting Improves Power for Rare Variant Association Studies

Abstract
:1. Introduction
2. Materials and Methods
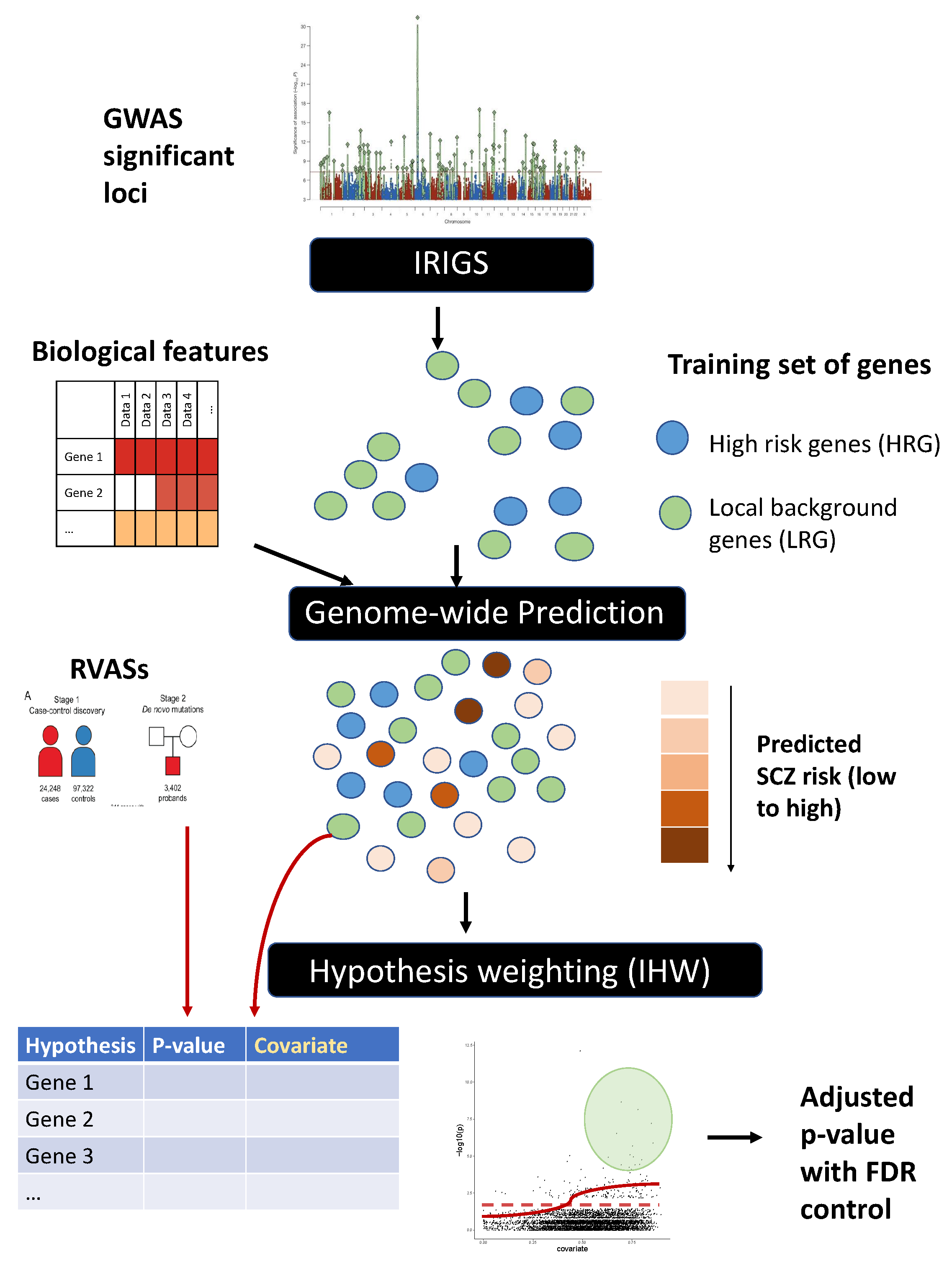
2.1. Method Overview
2.2. Obtain Training Set of Genes
2.3. Feature Pre-Processing
2.4. Model Training and Genome-Wide Prediction of SCZ Risk
2.5. Application of IHW for Hypothesis Weighting
2.6. The SCZ RVAS Data
2.7. The ASD RVAS Data
3. Results
3.1. Evaluation of Prediction Scores
3.2. Leverage Prediction as Covariates to Identify SCZ Risk Genes
3.3. Leverage Prediction as Covariates to Identify ASD Risk Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RVAS | rare variant association study |
| iRIGS | integrative risk gene selector |
| GWAS | genome-wide associations study |
| SCZ | schizophrenia |
| ASD | autism spectrum disorder |
| FDR | false discovery rate |
| FWER | family-wise error rate |
| PTV | protein-truncating variants |
| MPC | missense badness, PolyPhen-2, and constraint |
| AUC | area under the receiver-operating characteristic (ROC) curve |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Set | Short Description |
|---|---|
| FMRP-Darnel [55] | Fragile X mental retardation (FMRP) protein targets |
| RBFOX1 [56] | targets of RNA binding protein, fox-1 homolog 1 |
| PSD [57] | post synaptic genes |
| ECG [58] | evolutionary constrained genes |
| PRP [59] | genes related to presynaptic proteins |
| PRAZ [59] | genes in the presynaptic active zone |
| NMDAR [35] | components of the N-methyl-D-aspartate (NMDA) network |
| miR-137 targets [60] | miRNA-137 targets |
| GABA [61] | components of the GABA receptor complex |
| SYV [59] | synaptic vesicles |
| ARC | neuronal activity-regulated cytoskeleton-associated proteins |
| CRF [62] | chromatin remodeling factors |
| mGluR5 [46] | components of the metabotropic glutamate receptor 5 complex |
| CCS [63] | calcium channel and signaling genes |
References
- Liu, D.J.; Peloso, G.M.; Zhan, X.; Holmen, O.L.; Zawistowski, M.; Feng, S.; Nikpay, M.; Auer, P.L.; Goel, A.; Zhang, H.; et al. Meta-analysis of gene-level tests for rare variant association. Nat. Genet. 2014, 46, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.J.; Xia, F.; Zou, J. Fast and covariate-adaptive method amplifies detection power in large-scale multiple hypothesis testing. Nat. Commun. 2019, 10, 3433. [Google Scholar] [CrossRef] [Green Version]
- Genovese, C.R.; Roeder, K.; Wasserman, L. False discovery control with p-value weighting. Biometrika 2006, 93, 509–524. [Google Scholar] [CrossRef]
- Roeder, K.; Wasserman, L. Genome-wide significance levels and weighted hypothesis testing. Stat. Sci. Rev. J. Inst. Math. Stat. 2009, 24, 398. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Kabesch, M.; Bouzigon, E.; Demenais, F.; Farrall, M.; Moffatt, M.F.; Lin, X.; Liang, L. Using eQTL weights to improve power for genome-wide association studies: A genetic study of childhood asthma. Front. Genet. 2013, 4, 103. [Google Scholar] [CrossRef] [Green Version]
- Ignatiadis, N.; Klaus, B.; Zaugg, J.B.; Huber, W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 2016, 13, 577–580. [Google Scholar] [CrossRef] [Green Version]
- Kichaev, G.; Bhatia, G.; Loh, P.R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 2019, 104, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Andreassen, O.A.; Thompson, W.K.; Schork, A.J.; Ripke, S.; Mattingsdal, M.; Kelsoe, J.R.; Kendler, K.S.; O’Donovan, M.C.; Rujescu, D.; Werge, T.; et al. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet. 2013, 9, e1003455. [Google Scholar] [CrossRef] [Green Version]
- Yurko, R.; G’Sell, M.; Roeder, K.; Devlin, B. A selective inference approach for false discovery rate control using multiomics covariates yields insights into disease risk. Proc. Natl. Acad. Sci. USA 2020, 117, 15028–15035. [Google Scholar] [CrossRef] [PubMed]
- Fortney, K.; Dobriban, E.; Garagnani, P.; Pirazzini, C.; Monti, D.; Mari, D.; Atzmon, G.; Barzilai, N.; Franceschi, C.; Owen, A.B.; et al. Genome-wide scan informed by age-related disease identifies loci for exceptional human longevity. PLoS Genet. 2015, 11, e1005728. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Bai, L.; Cui, B.; Wu, L.; Wang, L.; An, Z.; Ruan, S.; Yu, Y.; Zhang, X.; Chen, J. Leveraging biological and statistical covariates improves the detection power in epigenome-wide association testing. Genome Biol. 2020, 21, 88. [Google Scholar] [CrossRef] [PubMed]
- Korthauer, K.; Kimes, P.K.; Duvallet, C.; Reyes, A.; Subramanian, A.; Teng, M.; Shukla, C.; Alm, E.J.; Hicks, S.C. A practical guide to methods controlling false discoveries in computational biology. Genome Biol. 2019, 20, 118. [Google Scholar] [CrossRef] [Green Version]
- Ignatiadis, N.; Huber, W. Covariate powered cross-weighted multiple testing. arXiv 2017, arXiv:1701.05179. [Google Scholar] [CrossRef]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia. medRxiv 2020. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Loos, R.J. 15 years of genome-wide association studies and no signs of slowing down. Nat. Commun. 2020, 11, 5900. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, R.; Cheng, F.; Wei, Q.; Ji, Y.; Yang, H.; Zhong, X.; Tao, R.; Wen, Z.; Sutcliffe, J.S.; et al. A Bayesian framework that integrates multi-omics data and gene networks predicts risk genes from schizophrenia GWAS data. Nat. Neurosci. 2019, 22, 691–699. [Google Scholar] [CrossRef]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.; Farh, K.H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421. [Google Scholar]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.K. Random decision forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; Volume 1, pp. 278–282. [Google Scholar]
- Miller, J.A.; Ding, S.L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Pers, T.H.; Karjalainen, J.M.; Chan, Y.; Westra, H.J.; Wood, A.R.; Yang, J.; Lui, J.C.; Vedantam, S.; Gustafsson, S.; Esko, T.; et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 2015, 6, 5890. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018, 36, 70. [Google Scholar] [CrossRef]
- Fernández, A.; García, S.; Galar, M.; Prati, R.C.; Krawczyk, B.; Herrera, F. Learning from Imbalanced Data Sets; Springer: Cham, Switzerland, 2018; Volume 10. [Google Scholar]
- James, G.; Witten, D.; Hastie, T.; Tibshirani, R. An Introduction to Statistical Learning; Springer: New York, NY, USA, 2013; Volume 112. [Google Scholar]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Bonferroni, C. Teoria statistica delle classi e calcolo delle probabilita. Pubbl. Ist. Super. Sci. Econ. Commericiali Firenze 1936, 8, 3–62. [Google Scholar]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.R.; Anttila, V.; Xu, H.; Zang, C.; Farh, K.; et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015, 47, 1228. [Google Scholar] [CrossRef] [Green Version]
- The Schizophrenia Working Group of the Psychiatric Genomics Consortium; Ripke, S.; Walters, J.T.; O’Donovan, M.C. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv 2020. [Google Scholar] [CrossRef]
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’dushlaine, C.; Chambert, K.; Bergen, S.E.; Kähler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callan, M.A.; Zarnescu, D.C. Heads-up: New roles for the fragile X mental retardation protein in neural stem and progenitor cells. Genesis 2011, 49, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Roeder, K.; Devlin, B.; Wasserman, L. Improving power in genome-wide association studies: Weights tip the scale. Genet. Epidemiol. Off. Publ. Int. Genet. Epidemiol. Soc. 2007, 31, 741–747. [Google Scholar] [CrossRef]
- Egbujo, C.N.; Sinclair, D.; Hahn, C.G. Dysregulations of synaptic vesicle trafficking in schizophrenia. Curr. Psychiatry Rep. 2016, 18, 77. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Sebat, J. CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Cell 2012, 148, 1223–1241. [Google Scholar] [CrossRef] [Green Version]
- Vergult, S.; Dheedene, A.; Meurs, A.; Faes, F.; Isidor, B.; Janssens, S.; Gautier, A.; Le Caignec, C.; Menten, B. Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur. J. Hum. Genet. 2015, 23, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, H.A.; Gerring, Z.; Hübel, C.; Middeldorp, C.M.; Derks, E.M.; Breen, G. Using genetic drug-target networks to develop new drug hypotheses for major depressive disorder. Transl. Psychiatry 2019, 9, 117. [Google Scholar] [CrossRef] [Green Version]
- Shimamoto, C.; Ohnishi, T.; Maekawa, M.; Watanabe, A.; Ohba, H.; Arai, R.; Iwayama, Y.; Hisano, Y.; Toyota, T.; Toyoshima, M.; et al. Functional characterization of FABP3, 5 and 7 gene variants identified in schizophrenia and autism spectrum disorder and mouse behavioral studies. Hum. Mol. Genet. 2014, 23, 6495–6511. [Google Scholar] [CrossRef] [Green Version]
- Tsavou, A.; Curtis, D. In-silico investigation of coding variants potentially affecting the functioning of the glutamatergic N-methyl-D-aspartate receptor in schizophrenia. Psychiatr. Genet. 2019, 29, 44–50. [Google Scholar] [CrossRef]
- Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; Witte, J.S.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984. [Google Scholar] [PubMed] [Green Version]
- Burbach, J.P.H.; Van der Zwaag, B. Contact in the genetics of autism and schizophrenia. Trends Neurosci. 2009, 32, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; McClellan, J.M.; McCarthy, S.E.; Addington, A.M.; Pierce, S.B.; Cooper, G.M.; Nord, A.S.; Kusenda, M.; Malhotra, D.; Bhandari, A.; et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008, 320, 539–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; Senman, L.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319. [Google Scholar] [PubMed] [Green Version]
- Gilbert, J.; Man, H.Y. Fundamental elements in autism: From neurogenesis and neurite growth to synaptic plasticity. Front. Cell. Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, R.; Pinyol, R.; Reichenbach, N.; Custer, L.; Klingensmith, J.; Kessels, M.M.; Qualmann, B. Cordon-bleu is an actin nucleation factor and controls neuronal morphology. Cell 2007, 131, 337–350. [Google Scholar] [CrossRef] [Green Version]
- Griswold, A.J.; Ma, D.; Cukier, H.N.; Nations, L.D.; Schmidt, M.A.; Chung, R.H.; Jaworski, J.M.; Salyakina, D.; Konidari, I.; Whitehead, P.L.; et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 2012, 21, 3513–3523. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.H.; Reutiman, T.J.; Folsom, T.D.; Thuras, P.D. GABA A receptor downregulation in brains of subjects with autism. J. Autism Dev. Disord. 2009, 39, 223. [Google Scholar] [CrossRef] [Green Version]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Loke, Y.J.; Hannan, A.J.; Craig, J.M. The role of epigenetic change in autism spectrum disorders. Front. Neurol. 2015, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Lei, L.; Fithian, W. Adapt: An interactive procedure for multiple testing with side information. arXiv 2016, arXiv:1609.06035. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyn-Vanhentenryck, S.M.; Mele, A.; Yan, Q.; Sun, S.; Farny, N.; Zhang, Z.; Xue, C.; Herre, M.; Silver, P.A.; Zhang, M.Q.; et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014, 6, 1139–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayés, À.; Van De Lagemaat, L.N.; Collins, M.O.; Croning, M.D.; Whittle, I.R.; Choudhary, J.S.; Grant, S.G. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011, 14, 19–21. [Google Scholar] [CrossRef] [Green Version]
- Samocha, K.E.; Robinson, E.B.; Sanders, S.J.; Stevens, C.; Sabo, A.; McGrath, L.M.; Kosmicki, J.A.; Rehnström, K.; Mallick, S.; Kirby, A.; et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014, 46, 944–950. [Google Scholar] [CrossRef]
- Pirooznia, M.; Wang, T.; Avramopoulos, D.; Valle, D.; Thomas, G.; Huganir, R.L.; Goes, F.S.; Potash, J.B.; Zandi, P.P. SynaptomeDB: An ontology-based knowledgebase for synaptic genes. Bioinformatics 2012, 28, 897–899. [Google Scholar] [CrossRef] [Green Version]
- Ripke, S.; Sanders, A.R.; Kendler, K.S.; Levinson, D.F.; Sklar, P.; Holmans, P.A.; Lin, D.Y.; Duan, J.; Ophoff, R.A.; Andreassen, O.A.; et al. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011, 43, 969. [Google Scholar]
- Pocklington, A.J.; Rees, E.; Walters, J.T.; Han, J.; Kavanagh, D.H.; Chambert, K.D.; Holmans, P.; Moran, J.L.; McCarroll, S.A.; Kirov, G.; et al. Novel findings from CNVs implicate inhibitory and excitatory signaling complexes in schizophrenia. Neuron 2015, 86, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- Shipra, A.; Chetan, K.; Rao, M. CREMOFAC—A database of chromatin remodeling factors. Bioinformatics 2006, 22, 2940–2944. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.S.; Haupt, A.; Bildl, W.; Schindler, J.; Knaus, H.G.; Meissner, M.; Rammner, B.; Striessnig, J.; Flockerzi, V.; Fakler, B.; et al. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14950–14957. [Google Scholar] [CrossRef] [Green Version]







| Gene Set | OR | p-Value |
|---|---|---|
| FMRP-Darnel (832) | 14.23 | |
| RBFOX1 (556) | 11.14 | |
| PSD (1444) | 5.15 | |
| ECG (998) | 5.38 | |
| PRP (336) | 5.06 | |
| PRAZ (209) | 5.87 | |
| NMDAR (59) | 18.17 | |
| miR-137 targets (281) | 4.32 | |
| GABA (18) | 46.06 | |
| SYV (107) | 4.32 | |
| ARC (25) | 13.82 | |
| CRF (56) | 5.54 | |
| mGluR5 (37) | 6.28 | |
| CCS (73) | 3.72 |
| Method | 0.05 | 0.1 | 0.2 | 0.3 |
|---|---|---|---|---|
| IHW-BRAINSPAN | 29 | 34 | 51 | 63 |
| IHW-FANTOM5 | 30 | 37 | 54 | 64 |
| IHW-DEPICT | 30 | 36 | 57 | 93 |
| IHW-LAKE | 30 | 35 | 55 | 97 |
| IHW-ensemble | 30 | 38 | 59 | 134 |
| IHW-shuffled ensemble d | 31 | 31 | 48 | 57 |
| BH | 31 | 33 | 46 | 64 |
| Method | 0.05 | 0.1 | 0.2 | 0.3 |
|---|---|---|---|---|
| IHW-BRAINSPAN | 89 | 142 | 294 | 477 |
| IHW-FANTOM5 | 98 | 119 | 207 | 363 |
| IHW-DEPICT | 107 | 158 | 329 | 495 |
| IHW-LAKE | 105 | 149 | 287 | 439 |
| IHW-ensemble | 112 | 176 | 323 | 658 |
| IHW-shuffled ensemble d | 79 | 95 | 151 | 201 |
| BH | 76 | 99 | 143 | 199 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Chen, R.; Wang, Q.; Wei, Q.; Tao, R.; Li, B. Leveraging Gene-Level Prediction as Informative Covariate in Hypothesis Weighting Improves Power for Rare Variant Association Studies. Genes 2022, 13, 381. https://doi.org/10.3390/genes13020381
Ji Y, Chen R, Wang Q, Wei Q, Tao R, Li B. Leveraging Gene-Level Prediction as Informative Covariate in Hypothesis Weighting Improves Power for Rare Variant Association Studies. Genes. 2022; 13(2):381. https://doi.org/10.3390/genes13020381
Chicago/Turabian StyleJi, Ying, Rui Chen, Quan Wang, Qiang Wei, Ran Tao, and Bingshan Li. 2022. "Leveraging Gene-Level Prediction as Informative Covariate in Hypothesis Weighting Improves Power for Rare Variant Association Studies" Genes 13, no. 2: 381. https://doi.org/10.3390/genes13020381
APA StyleJi, Y., Chen, R., Wang, Q., Wei, Q., Tao, R., & Li, B. (2022). Leveraging Gene-Level Prediction as Informative Covariate in Hypothesis Weighting Improves Power for Rare Variant Association Studies. Genes, 13(2), 381. https://doi.org/10.3390/genes13020381