
Genetic Structure of the Root Vole Microtus oeconomus: Resistance of the Habitat Specialist to the Natural Fragmentation of Preferred Moist Habitats

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
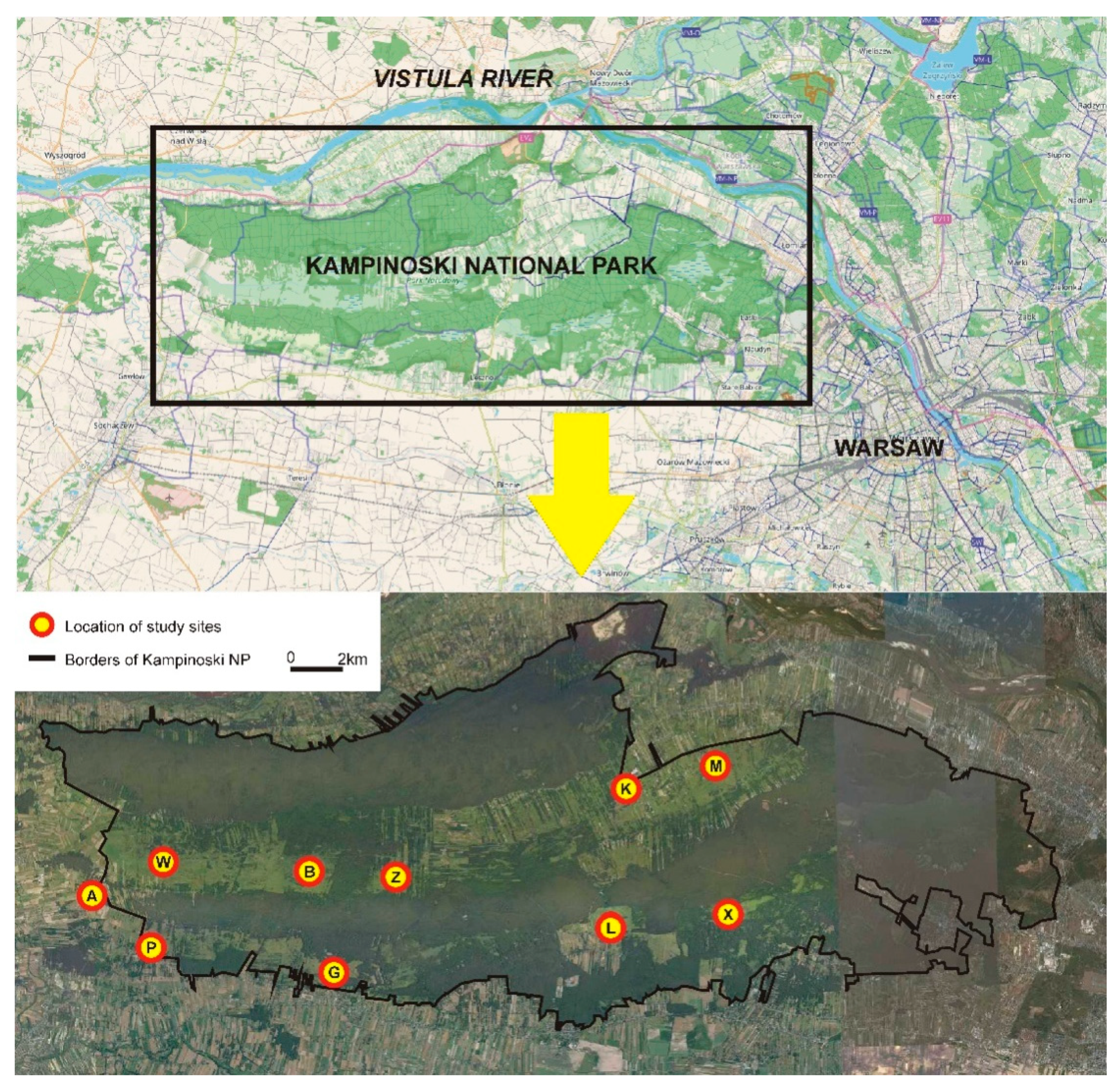
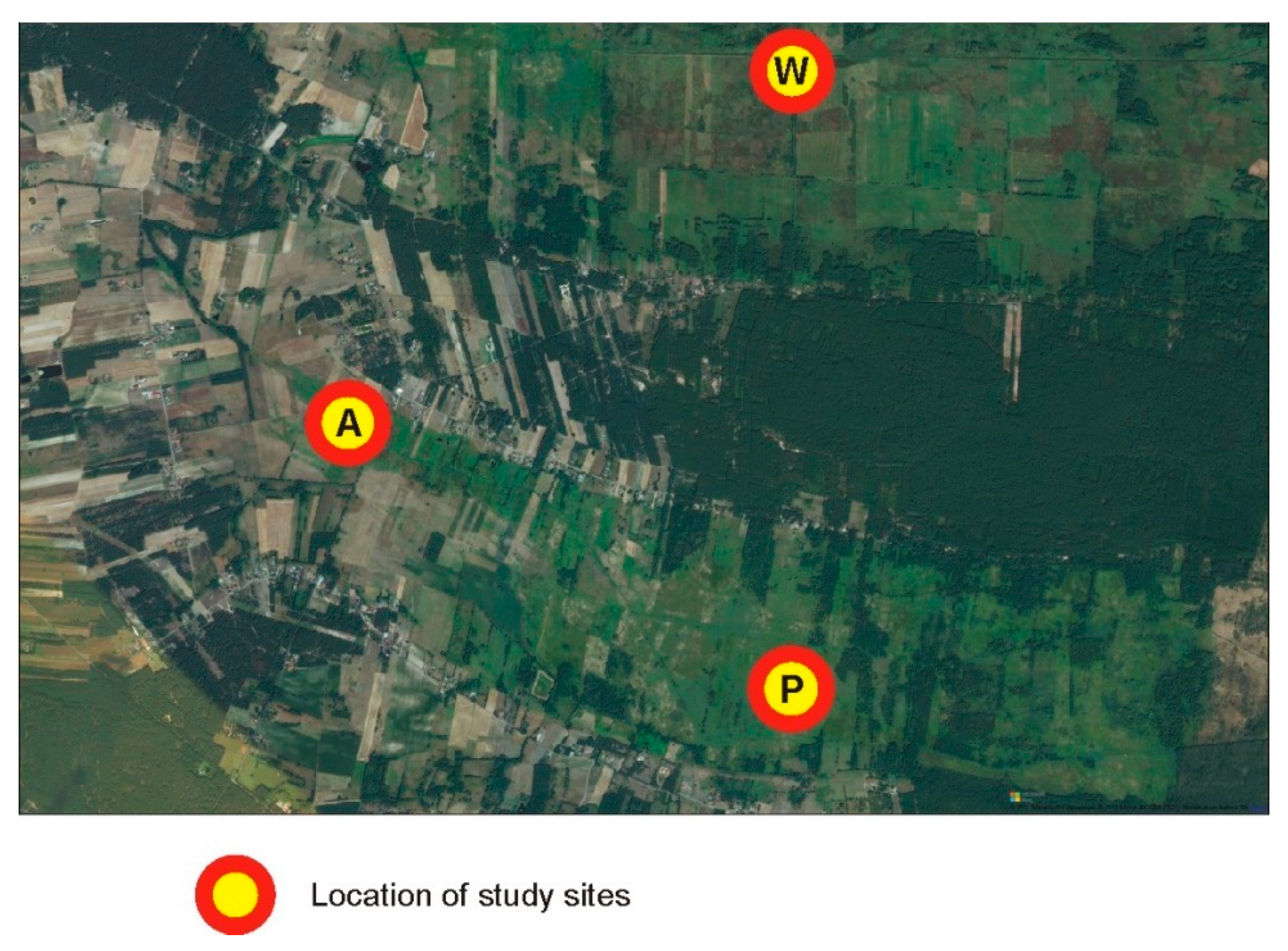
2.1. Study Area and Tissue Sampling
2.2. Small Mammal Trapping
2.3. Microsatellite Genotyping
2.4. Analyses of Genetic Structure and Variability
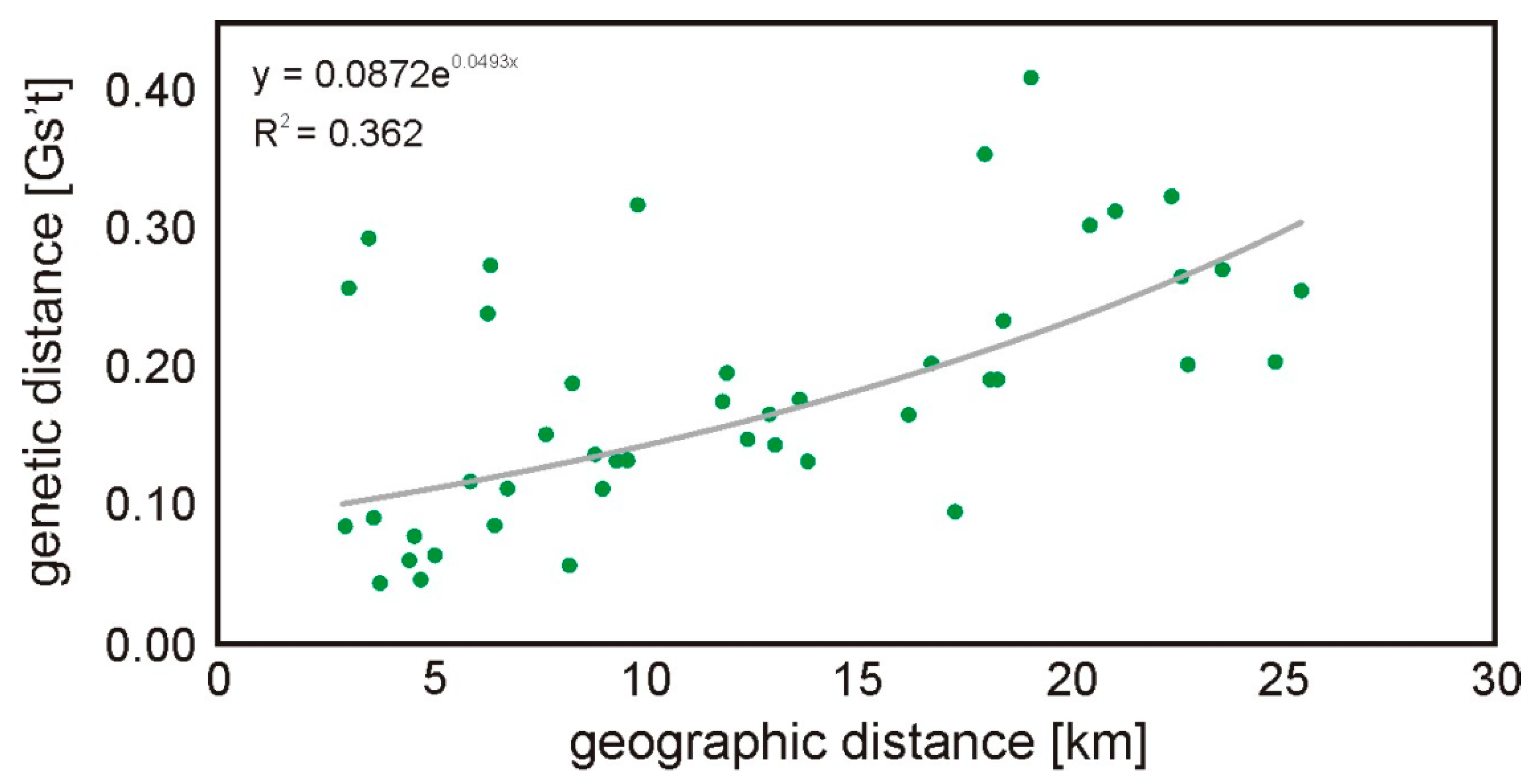
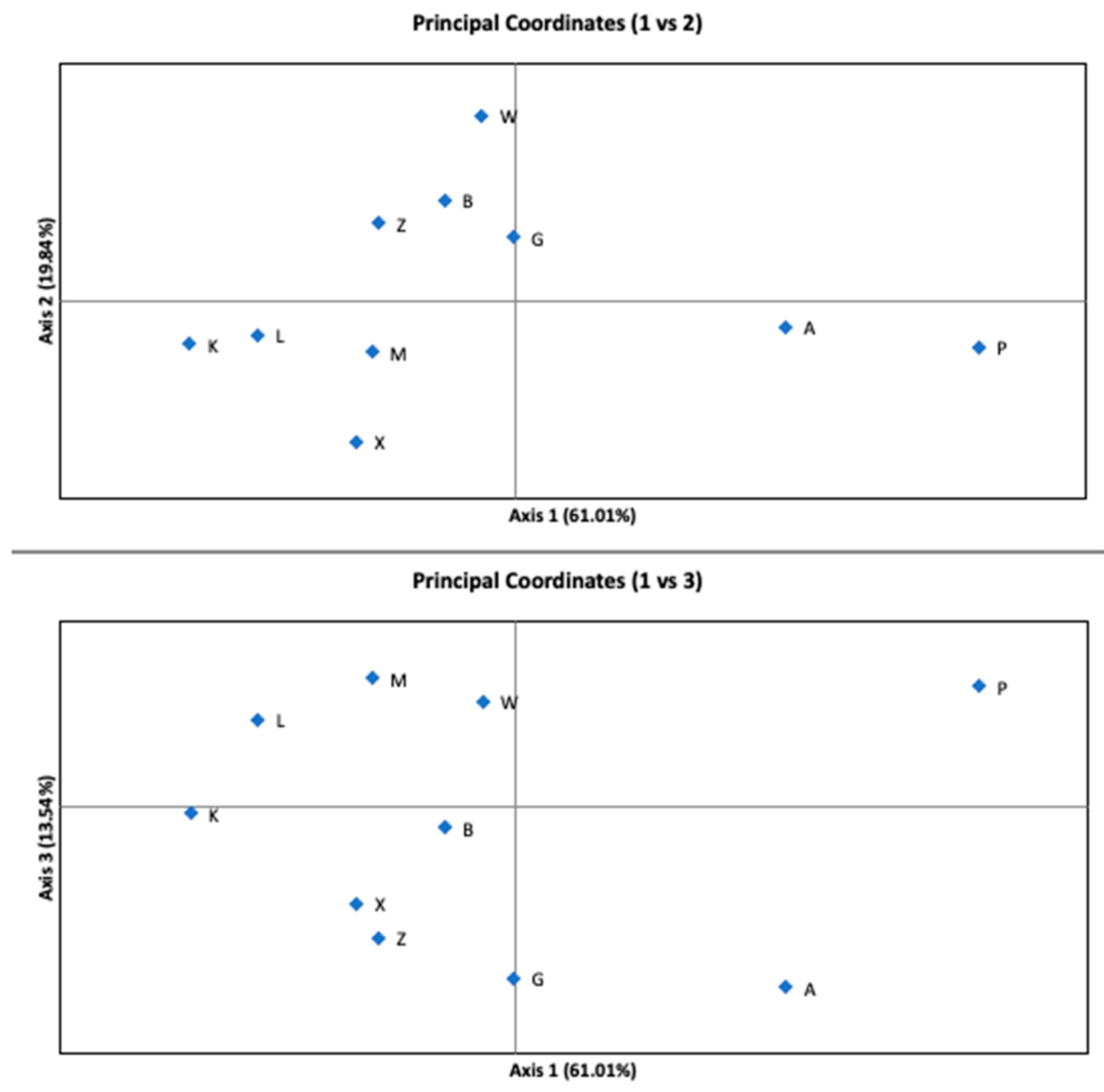
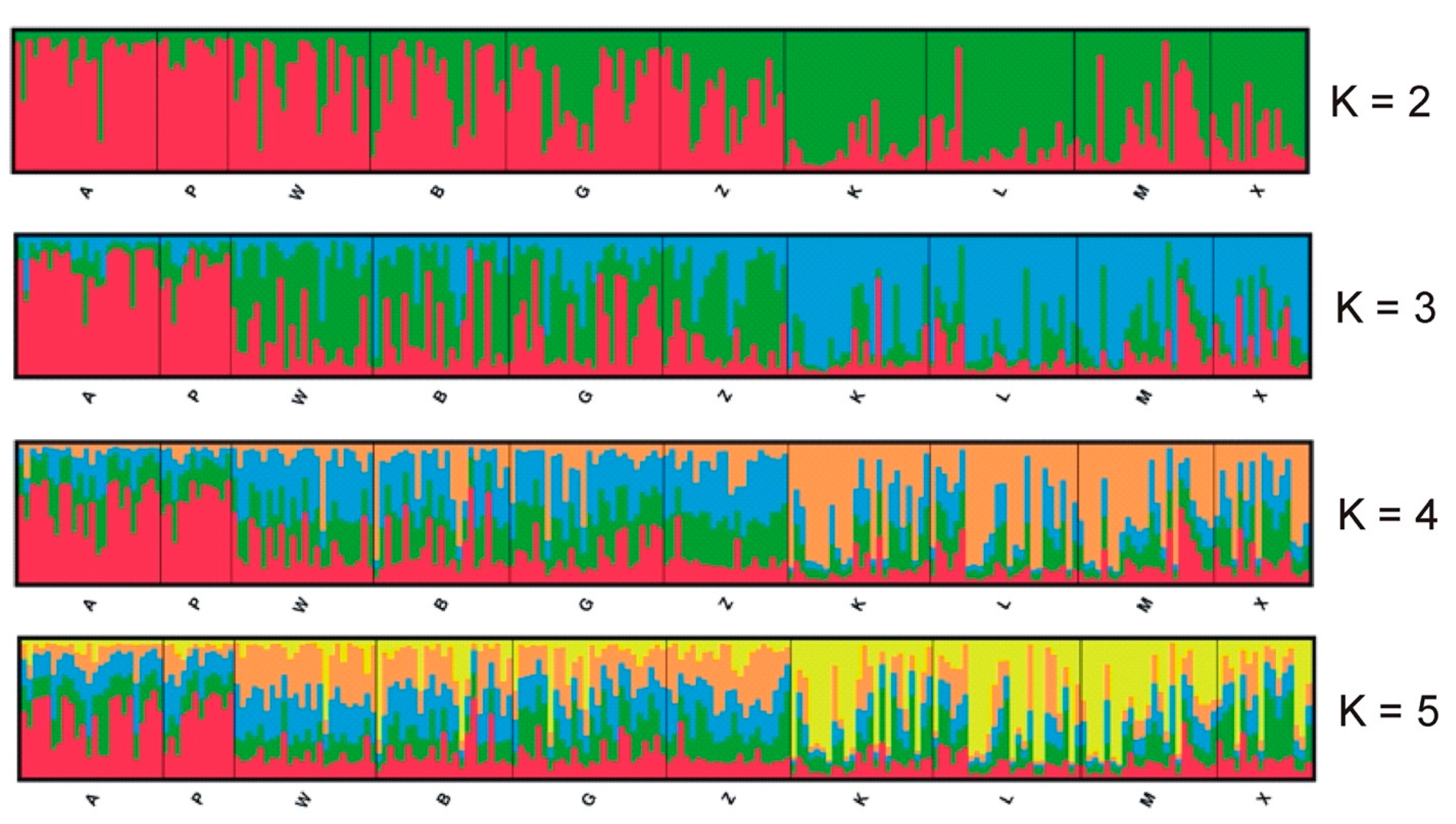
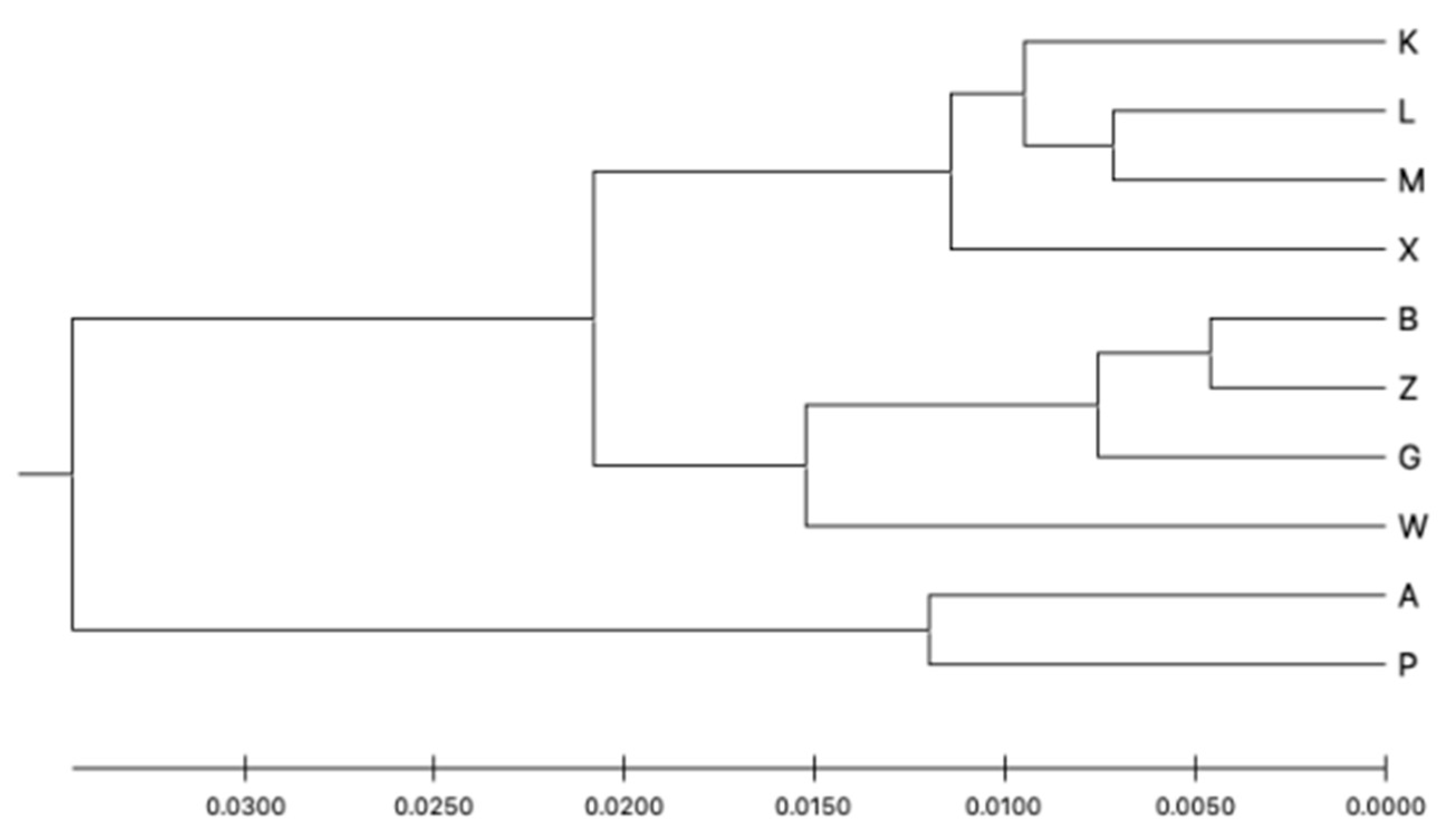
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klimkowska, A.; Dzierża, P.; Grootjans, A.P.; Kotowski, W.; VAN Diggelen, R. Prospects of fen restoration in relation to changing land use—An example from central Poland. Landsc. Urban Plan. 2010, 97, 249–257. [Google Scholar] [CrossRef]
- Junk, W.J.; An, S.; Finlayson, C.M. Current state of knowledge regarding the world’s wetlands and their future under global climate change: A synthesis. Aquat. Sci. 2013, 75, 151–167. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.P. Wetland loss and biodiversity conservation. Conserv. Biol. 2000, 14, 314–317. [Google Scholar] [CrossRef] [Green Version]
- Hanski, I. Metapopulation dynamics. Nature 2018, 396, 41–49. [Google Scholar] [CrossRef]
- Young, A.G.; Clarke, G.M. Genetics, Demography and Viability of Fragmented Populations; Cambridge University Press: Cambridge, UK, 2000; pp. 1–456. [Google Scholar]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Keyghobadi, N. The genetic implications of habitat fragmentation in animals. Can. J. Zool. 2007, 85, 1049–1064. [Google Scholar] [CrossRef]
- Loew, S.S.; Williams, D.F.; Ralls, K.; Pilgrim, K.; Fleischer, R.C. Population structure and genetic variation in the endangered Giant Kangaroo Rat (Dipodomysingens). Conserv. Genet. 2005, 6, 495–510. [Google Scholar] [CrossRef]
- Wood, A.R.; Gardner, J.P.A. Small spatial scale population genetic structure in two limpet species endemic to the Kermadec Islands, New Zealand. Mar. Ecol. Prog. Ser. 2007, 349, 159–170. [Google Scholar] [CrossRef]
- Gerlach, G.; Musolf, K. Fragmentation of landscape as a cause for genetic subdivision in bank voles. Conserv. Biol. 2000, 14, 1066–1074. [Google Scholar] [CrossRef]
- Ratkiewicz, M.; Borkowska, A. Genetic structure is influence by environmental barriers: Empirical evidence from the common vole Microtus arvalis populations. Acta Theriol. 2006, 51, 337–344. [Google Scholar] [CrossRef]
- Biedrzycka, A.; Konopiński, M.K. Genetic variability and the effect of habitat fragmentation in spotted suslik Spermophilus suslicus populations from two different regions. Conserv. Genet. 2008, 9, 1211–1221. [Google Scholar] [CrossRef]
- Wilson, D.E.; Reeder, D.M. (Eds.) Mammal Species of the World. In A Taxonomic and Geographic Reference, 3rd ed.; Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]
- Aars, J.; Ims, R.A. Population dynamic and genetic consequences of spatial density-dependent dispersal in patchy populations. Am. Nat. 2000, 155, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Hulejová Sládkovičová, V.; Dąbrowski, M.J.; Žak, D.; Miklós, P.; Gubányi, A.; La Haye, M.J.J.; Bekker, D. Genetic variability of the cold-tolerant Microtus oeconomus subspecies left behind retreating glaciers. Mamm. Biol. 2018, 88, 85–93. [Google Scholar] [CrossRef]
- Galbreath, K.; Cook, J. Genetic consequences of Pleistocene glaciations for the tundra vole (Microtus oeconomus) in Beringia. Mol. Ecol. 2004, 13, 135–148. [Google Scholar] [CrossRef]
- Brunhoff, C.; Yoccoz, N.G.; Ims, R.A.; Jaarola, M. Glacial survival or late glacial colonization? Phylogeography of the root vole (Microtus oeconomus) in north-west Norway. J. Biogeogr. 2006, 33, 2136–2144. [Google Scholar] [CrossRef]
- Jancewicz, E.; Falkowska, E. The effect of Pleistocene glacial morphogenesis on the genetic structure of the humid- and cold-tolerant root vole Microtus oeconomus (Rodentia, Cricetidae) in Poland, central Europe. Quat. Res. 2020, 93, 225–242. [Google Scholar] [CrossRef]
- Sałata-Pilacińska, B. The southern range of the root vole in Poland. Acta Theriol. 1990, 35, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Tast, J. Microtus oeconomus (Pallas, 1776). In Handbuch der Säugetiere Europas; Niethammer, J., Krapp, F., Eds.; Akademische Verlagsgesellschaft: Wiesbade, Germany, 1982; pp. 145–148. [Google Scholar]
- Horváth, G.F.; Herczeg, R. Site occupancy response to natural and anthropogenic disturbances of root vole: Conservation problem of a vulnerable relict subspecies. J. Nat. Conserv. 2013, 21, 350–358. [Google Scholar] [CrossRef]
- Van de Zande, L.; Van Apeldoorn, R.C.; Blijdenstein, A.F.; de Jong, D.; van Delden, W.; Bijlsma, R. Microsatellite analysis of population structure and genetic differentiation within and between populations of the root vole, Microtus oeconomus in the Netherlands. Mol. Ecol. 2000, 9, 1651–1656. [Google Scholar] [CrossRef] [Green Version]
- Kelemen, K.A.; Urzi, F.; Buzan, E.; Horváth, G.F.; Tulis, F.; Baláž, I. Genetic variability and conservation of the endangered Pannonian root vole in fragmented habitats of an agricultural landscape. Nat. Conserv. 2021, 43, 167. [Google Scholar] [CrossRef]
- Goldstein, D.B.; Schlotterer, C. Microsatellites: Evolution and Applications; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Storfer, A.; Murphy, M.A.; Spear, S.F.; Holderegger, R.; Waits, L.P. Landscape genetics: Where are we now? Mol. Ecol. 2010, 19, 3496–3514. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Yoshinaga, Y.; Saitoh, T.; Abe, S.; Iida, H.; Yoshida, M.C. Polymorphic microsatellite DNA markers in the field vole Microtus montebelli. Mol. Ecol. 1999, 8, 157–168. [Google Scholar]
- Papp, T.; Gubanyi, A.; Racz, G. Establishing microsatellite analysis for locally endangered populations of root vole (Microtus oeconomus). Acta Zool. Hung. 2000, 46, 259–264. [Google Scholar]
- Jaarola, M.; Ratkiewicz, M.; Ashford, R.T.; Brunhoff, C.; Borkowska, A. Isolation and characterization of polymorphic microsatellite loci in the field vole, Microtus agrestis, and their cross-utility in the common vole, Microtus arvalis. Mol. Ecol. Notes 2007, 7, 1029–1031. [Google Scholar] [CrossRef]
- Borkowska, A.; Borowski, Z.; Krysiuk, K. Multiple paternity in free-living root voles (Microtus oeconomus). Behav. Processes 2009, 82, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Czajkowska, M.; Borkowska, A.; Wieczorek, M.; Zub, K. Application of microsatellite markers developed for arvicoline species in a population genetic study of the root vole Microtus oeconomus. Acta Theriol. 2010, 55, 123–128. [Google Scholar] [CrossRef]
- Pilot, M.; Dąbrowski, M.J.; Jancewicz, E.; Schtickzelle, N.; Gliwicz, J. Temporally stablegenetic variability and dynamic kinship structure in a fluctuating population of the root vole Microtus oeconomus. Mol. Ecol. 2010, 19, 2800–2812. [Google Scholar] [CrossRef]
- Toth, B.; Khosravi, R.; Ashrafzadeh, M.R. Genetic Diversity and Structure of Common Carp (Cyprinus carpio L.) in the Centre of Carpathian Basin: Implications for Conservation. Genes 2020, 11, 1268. [Google Scholar] [CrossRef]
- Mihalik, B.; Frank, K.; Astuti, P.K. Population Genetic Structure of the Wild Boar (Sus scrofa) in the Carpathian Basin. Genes 2020, 11, 1194. [Google Scholar] [CrossRef] [PubMed]
- Abdelmanova, A.S.; Kharzinova, V.R.; Volkova, V.V. Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis. Genes 2020, 11, 940. [Google Scholar] [CrossRef]
- Paredes, G.F.; Yalta-Macedo, C.E.; Gutierrez, G.A. Genetic Diversity and Population Structure of Llamas (Lama glama) from the Camelid Germplasm Bank-Quimsachata. Genes 2020, 11, 541. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, R. (Ed.) Kampinoski Park Narodowy; tom I. Kampinoski Park Narodowy: Izabelin, Poland, 2003. [Google Scholar]
- Larbi, B.M.; Tircazes, A.; Feve, K.; Tudela, F.; Bolet, G. Reliability of non-invasive tissue sampling methods for DNA extraction in rabbits (Oryctolagus cuniculus). World Rabbit Sci. 2012, 20, 117–124. [Google Scholar]
- Goudet, J. FSTAT Version 1.2: A computer program to calculate F-statistics. J. Heredity 1995, 86, 485–486. [Google Scholar]
- Meirmans, P.G. GenoDive version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Res. 2020, 20, 1126–1131. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, P. A standardized genetic differentiation measure. Evolution 2005, 59, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research_an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, M.; Rousset, F. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J. Heredity 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A complete reimplementation of the Genepop software for Windows and Linux. Mol. Ecol. Res. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Nei, M.; Tajima, F.; Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef]
- Sneath, P.H.A.; Sokal, R.R. Numerical Taxonomy; Freeman: San Francisco, CA, USA, 1973. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Gen. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Eco. Res. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balčiauskas, L.; Balčiauskienė, L.; Baltrūnaitė, L. Root vole, Microtus oeconomus, in Lithuania: Changes in the distribution range. J. Vertebr. Biol. 2010, 59, 267–277. [Google Scholar] [CrossRef]
- Balčiauskas, L.; Balčiauskienė, L.; Stirkė, V. Mow the Grass at the Mouse’s Peril: Diversity of Small Mammals in Commercial Fruit Farms. Animals 2019, 9, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łopucki, R.; Mróz, I.; Klich, D.; Kitowski, I. Small mammals of xerothermic grasslands of south-eastern Poland. Ann. Warsaw Univ. of Life Sci.–SGGW Anim. Sci. 2018, 57, 257–267. [Google Scholar] [CrossRef]
- Odden, M.; Ims, R.A.; Støen, O.G.; Swenson, J.E.; Andreassen, H.P. Bears are simply voles writ large: Social structure determines the mechanisms of intrinsic population regulation in mammals. Oecologia 2014, 175, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lesiński, G.; Romanowski, J.; Gryz, J.; Olszewski, A.; Kowalski, M.; Krauze-Gryz, D.; Taowski, A. Small mammals of Kampinos National Park and its protection zone, as revealed by analyses of the diet of tawny owls Strix aluco Linnaeus, 1758. Fragm. Faunist. 2013, 56, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Leijs, R.; van Apeldoorn, R.C.; Bijlsma, R. Low genetic differentiation in north-west European populations of the locally endangered root vole, Microtus oeconomus. Biol. Conserv. 1999, 87, 39–48. [Google Scholar] [CrossRef]
- Stojak, J.; Wójcik, J.M.; Ruczyńska, I.; Searle, J.B.; McDevitt, A.D. Contrasting and congruent patterns of genetic structuring in two Microtus vole species using museum specimens. Mamm. Res. 2016, 61, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Gaines, M.S.; Diffendorfer, J.E.; Tamarin, R.H.; Whittam, T.S. The effects of habitat fragmentation on the genetic structure of small mammal populations. J. Hered. 1997, 88, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Łopucki, R.; Klich, D.; Kitowski, I.; Kiersztyn, A. Urban size effect on biodiversity: The need for a conceptual framework for the implementation of urban policy for small cities. Cities 2020, 98, 102590. [Google Scholar] [CrossRef]
- Łopucki, R.; Kitowski, I. How small cities affect the biodiversity of ground-dwelling mammals and the relevance of this knowledge in planning urban land expansion in terms of urban wildlife. Urban Ecosyst. 2017, 20, 933. [Google Scholar] [CrossRef] [Green Version]
- Clevenger, A.P.; Chruszcz, B.; Gunson, K.E. Spatial patterns and factors influencing small vertebrate fauna road-kill aggregations. Biol. Cons. 2002, 109, 15–26. [Google Scholar] [CrossRef]
- Coffin, A.W. From roadkill to road ecology: A review of the ecological effects of roads. J. Trans. Geogr. 2007, 15, 396–406. [Google Scholar] [CrossRef]
- Glista, D.J.; DeVault, T.L.; DeWoody, J.A. A review of mitigation measures for reducing wildlife mortality on roadways. Landsc. Urban Plan. 2009, 91, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gunson, K.E.; Mountrakis, G.; Quackenbush, L.J. Spatial wildlife-vehicle collision models: A review of current work and its application to transportation mitigation projects. J. Environ. Manag. 2011, 92, 1074–1082. [Google Scholar] [CrossRef]
- Jackson, N.D.; Fahrig, L. Relative effects of road mortality and decreased connectivity on population genetic diversity. Biol. Conserv. 2011, 144, 3143–3148. [Google Scholar] [CrossRef]
- Bąkowski, C.; Kozakiewicz, M. The effect of forest road on bank vole and yellow-necked mouse populations. Acta Theriol. 1988, 33, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Krauze-Gryz, D.; Gryz, J.; Goszczyński, J. Predation by domestic cats in rural areas of Central Poland: An assessment based on two methods. J. Zool. 2012, 288, 260–266. [Google Scholar] [CrossRef]
- Krauze-Gryz, D.; Żmihorski, M.; Gryz, J. Annual variation in prey composition of domestic cats in rural and urban environment. Urban Ecosyst. 2017, 20, 945–952. [Google Scholar] [CrossRef] [Green Version]
- Bjørnstad, O.N.; Andreassen, H.P.; Ims, R.A. Effects of habitat patchiness and connectivity on the spatial ecology of the root vole Microtus oeconomus. J. Anim. Ecol. 1998, 67, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef]
- Nei, M.; Maruyama, T.; Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Lambin, X.; Krebs, C. Can changes in female relatedness influence microtine population dynamics? Oikos 1991, 61, 126–132. [Google Scholar] [CrossRef]
- Coltman, D.W.; Pilkington, J.G.; Pemberton, J.M. Fine-scale genetic structure in a free-living ungulate population. Mol. Ecol. 2003, 12, 733–742. [Google Scholar] [CrossRef]
- Nussey, D.H.; Coltman, D.W.; Coulson, T. Rapidly declining fine-scale spatial genetic structure in female red deer. Mol. Ecol. 2005, 14, 3395–3405. [Google Scholar] [CrossRef] [PubMed]
- Kozakiewicz, M.; Gortat, T.; Kozakiewicz, A.; Barkowska, M. Effects of habitat fragmentation on four rodent species in a Polish farm landscape. Landsc. Ecol. 1999, 14, 391–400. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WEST | CENTRE | EAST | ||||
|---|---|---|---|---|---|---|
| Northern Strip | Study site | W | B | Z | K | M |
| Number of samples | 24 | 23 | 21 | 24 | 23 | |
| Southern Strip | Study site | A | P | G | L | X |
| Number of samples | 24 | 12 | 26 | 25 | 16 | |
| Location | Na | AR | Ho | He | HWE | Gis | |
|---|---|---|---|---|---|---|---|
| p-Value | ±SE | ||||||
| A | 6.89 | 5.53 | 0.446 | 0.751 | 0.000 | 0.000 | 0.406 *** |
| P | 6.22 | 5.89 | 0.437 | 0.755 | 0.000 | 0.000 | 0.422 *** |
| W | 7.56 | 6.00 | 0.656 | 0.781 | 0.000 | 0.000 | 0.160 *** |
| B | 8.44 | 6.47 | 0.662 | 0.806 | 0.000 | 0.000 | 0.178 *** |
| G | 7.44 | 6.04 | 0.677 | 0.792 | 0.000 | 0.000 | 0.146 *** |
| Z | 6.89 | 6.09 | 0.564 | 0.822 | 0.000 | 0.000 | 0.314 *** |
| K | 6.44 | 5.40 | 0.628 | 0.749 | 0.000 | 0.000 | 0.162 *** |
| L | 6.89 | 5.51 | 0.703 | 0.771 | 0.011 | 0.007 | 0.088 ** |
| M | 7.56 | 5.94 | 0.695 | 0.780 | 0.000 | 0.000 | 0.108 ** |
| X | 6.33 | 5.40 | 0.513 | 0.762 | 0.000 | 0.000 | 0.326 *** |
| A | B | G | K | L | M | P | W | X | Z | |
|---|---|---|---|---|---|---|---|---|---|---|
| A | -- | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.036 | 0.000 | 0.000 | 0.000 |
| B | 0.046 | -- | 0.002 | 0.000 | 0.000 | 0.003 | 0.000 | 0.000 | 0.000 | 0.082 |
| G | 0.029 | 0.020 | -- | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.064 |
| K | 0.086 | 0.041 | 0.045 | -- | 0.006 | 0.004 | 0.000 | 0.000 | 0.007 | 0.000 |
| L | 0.080 | 0.035 | 0.042 | 0.017 | -- | 0.017 | 0.000 | 0.000 | 0.034 | 0.000 |
| M | 0.066 | 0.022 | 0.045 | 0.021 | 0.014 | -- | 0.000 | 0.000 | 0.005 | 0.001 |
| P | 0.024 | 0.066 | 0.060 | 0.111 | 0.092 | 0.069 | -- | 0.000 | 0.000 | 0.000 |
| W | 0.066 | 0.027 | 0.036 | 0.060 | 0.048 | 0.049 | 0.075 | -- | 0.000 | 0.000 |
| X | 0.055 | 0.048 | 0.041 | 0.024 | 0.016 | 0.029 | 0.086 | 0.067 | -- | 0.001 |
| Z | 0.046 | 0.009 | 0.010 | 0.031 | 0.030 | 0.029 | 0.073 | 0.029 | 0.033 | -- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łopucki, R.; Mróz, I.; Nowak-Życzyńska, Z.; Perlińska-Teresiak, M.; Owadowska-Cornil, E.; Klich, D. Genetic Structure of the Root Vole Microtus oeconomus: Resistance of the Habitat Specialist to the Natural Fragmentation of Preferred Moist Habitats. Genes 2022, 13, 434. https://doi.org/10.3390/genes13030434
Łopucki R, Mróz I, Nowak-Życzyńska Z, Perlińska-Teresiak M, Owadowska-Cornil E, Klich D. Genetic Structure of the Root Vole Microtus oeconomus: Resistance of the Habitat Specialist to the Natural Fragmentation of Preferred Moist Habitats. Genes. 2022; 13(3):434. https://doi.org/10.3390/genes13030434
Chicago/Turabian StyleŁopucki, Rafał, Iwona Mróz, Zuzanna Nowak-Życzyńska, Magdalena Perlińska-Teresiak, Edyta Owadowska-Cornil, and Daniel Klich. 2022. "Genetic Structure of the Root Vole Microtus oeconomus: Resistance of the Habitat Specialist to the Natural Fragmentation of Preferred Moist Habitats" Genes 13, no. 3: 434. https://doi.org/10.3390/genes13030434
APA StyleŁopucki, R., Mróz, I., Nowak-Życzyńska, Z., Perlińska-Teresiak, M., Owadowska-Cornil, E., & Klich, D. (2022). Genetic Structure of the Root Vole Microtus oeconomus: Resistance of the Habitat Specialist to the Natural Fragmentation of Preferred Moist Habitats. Genes, 13(3), 434. https://doi.org/10.3390/genes13030434