
A Survey of Multigenic Protein-Altering Variant Frequency in Familial Exudative Vitreo-Retinopathy (FEVR) Patients by Targeted Sequencing of Seven FEVR-Linked Genes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
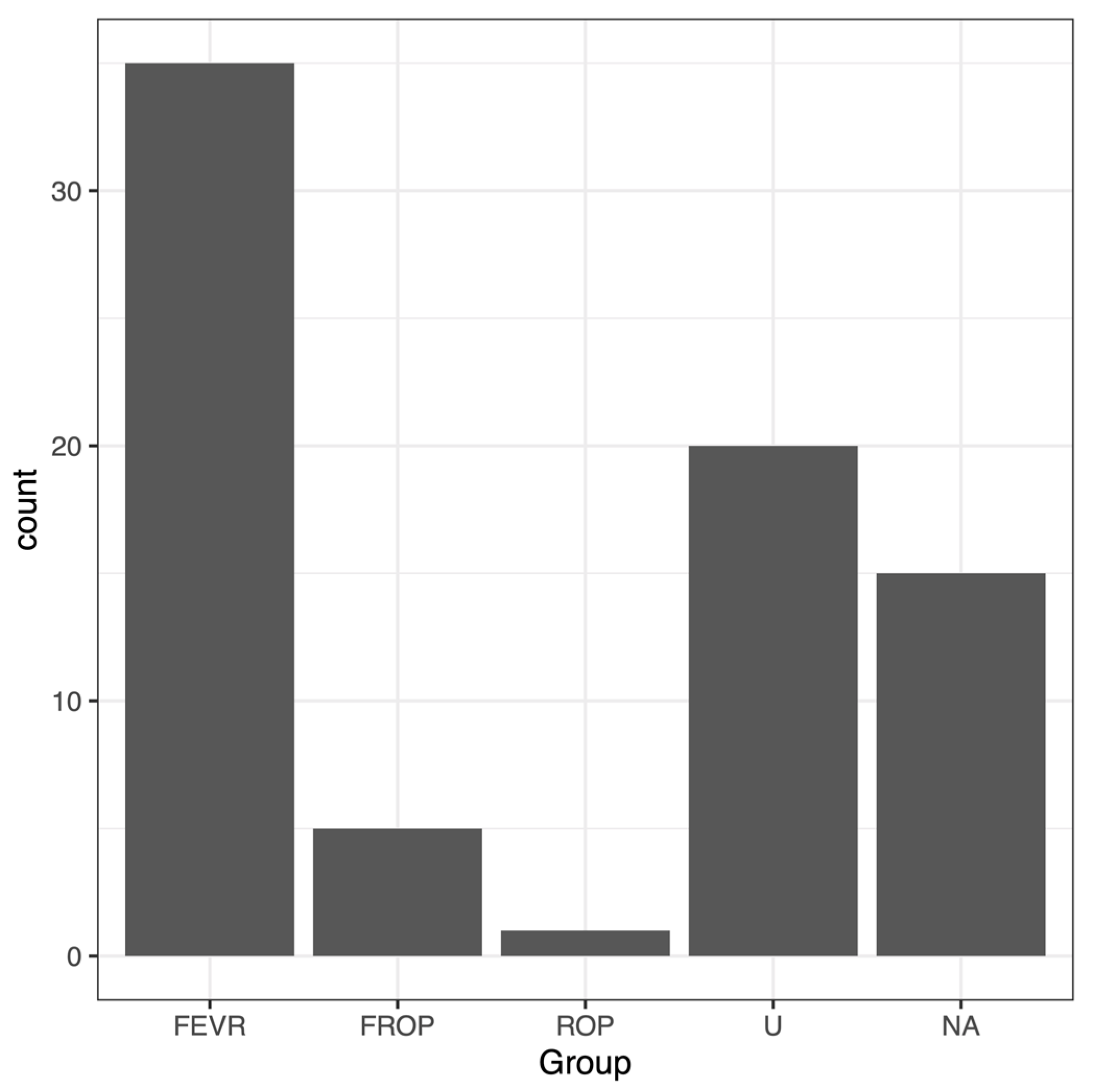
2.1. IRB Protocol and Test Group Characteristics
2.2. Sample Storage and Handling
2.3. Extraction of Genomic DNA
2.4. AmpliSeq Targeted Library Design In Silico
2.5. AmpliSeq Targeted Library Preparation
2.6. Sequencing on the iSeq-100 System
2.7. Bioinformatics Data and Analysis
3. Results
3.1. Sequencing Data Quality and Performance
3.2. Total Variant Detection Results
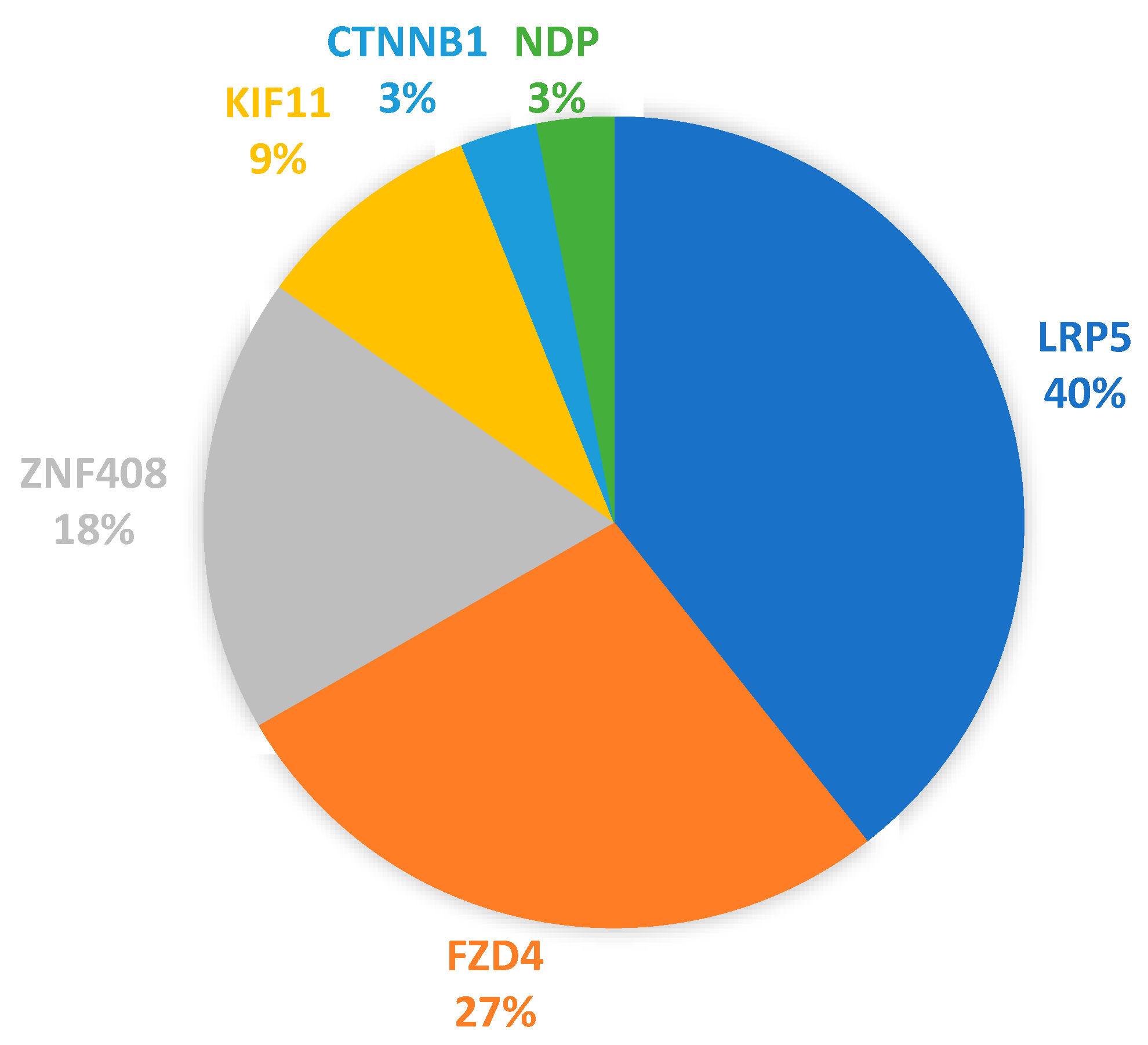
3.3. Protein-Altering Variants in Cohort of 76 FEVR Subjects and Relatives
3.4. Multiple Variants and Multigenic Variants
4. Discussion
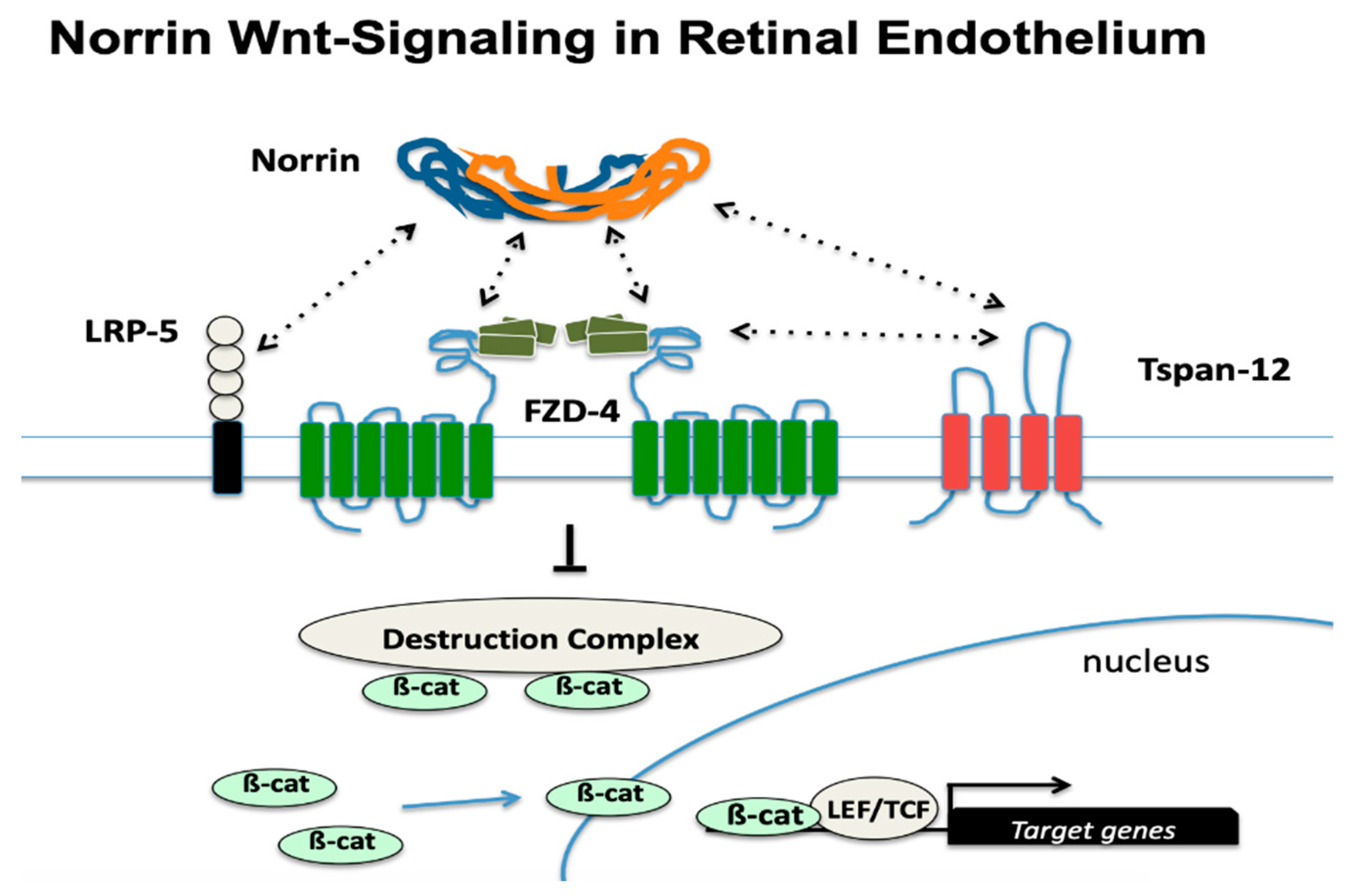
4.1. FEVR Phenotype and Genes
4.2. Frequency of Multigenic Protein-Altering Variants
4.3. Known and Novel Pathogenic Variants
4.4. Sequencing Approach
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drenser, K.A.; Dailey, W.; Vinekar, A.; Dalal, K.; Capone, A.; Trese, M.T. Clinical Presentation and Genetic Correlation of Patients With Mutations Affecting the FZD4 Gene. Arch. Ophthalmol. 2009, 127, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauqeer, Z.; Yonekawa, Y. Familial Exudative Vitreoretinopathy: Pathophysiology, Diagnosis, and Management. Asia Pacific J. Ophthalmol. 2018, 7, 176–182. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, J.; Yang, J.; Zhang, X.; Zhang, Q.; Zhao, P. Prenatal diagnosis of familial exudative vitreoretinopathy and Norrie disease. Mol. Genet. Genom. Med. 2018, 7, e00503. [Google Scholar] [CrossRef] [PubMed]
- Criswick, V.G.; Schepens, C.L. Familial exudative vitreoretinopathy. Am. J. Ophthalmol. 1969, 68, 578–594. [Google Scholar] [CrossRef]
- Shastry, B.S.; Trese, M.T. Cosegregation of two unlinked mutant alleles in some cases of autosomal dominant familial exudative vitreoretinopathy. Eur. J. Hum. Genet. 2003, 12, 79–82. [Google Scholar] [CrossRef]
- Li, Y.; Peng, J.; Li, J.; Zhang, Q.; Li, J.; Zhang, X.; Fei, P.; She, K.; Zhao, P. The characteristics of digenic familial exudative vitreoretinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 2149–2156. [Google Scholar] [CrossRef]
- Wang, Y.; Cho, C.; Williams, J.; Smallwood, P.M.; Zhang, C.; Junge, H.J.; Nathans, J. Interplay of the Norrin and Wnt7a/Wnt7b signaling systems in blood–brain barrier and blood–retina barrier development and maintenance. Proc. Natl. Acad. Sci. USA 2018, 115, E11827–E11836. [Google Scholar] [CrossRef] [Green Version]
- Collin, R.W.J.; Nikopoulos, K.; Dona, M.; Gilissen, C.; Hoischen, A.; Boonstra, F.N.; Poulter, J.A.; Kondo, H.; Berger, W.; Toomes, C.; et al. ZNF408 is mutated in familial exudative vitreoretinopathy and is crucial for the development of zebrafish retinal vasculature. Proc. Natl. Acad. Sci. USA 2013, 110, 9856–9861. [Google Scholar] [CrossRef] [Green Version]
- Robitaille, J.M.; Gillett, R.M.; LeBlanc, M.A.; Gaston, D.; Nightingale, M.; Mackley, M.P.; Parkash, S.; Hathaway, J.; Thomas, A.; Ells, A.; et al. Phenotypic Overlap Between Familial Exudative Vitreoretinopathy and Microcephaly, Lymphedema, and Chorioretinal Dysplasia Caused byKIF11Mutations. JAMA Ophthalmol. 2014, 132, 1393–1399. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Xiao, X.; Li, S.; Jia, X.; Guo, X.; Zhang, Q. KIF11mutations are a common cause of autosomal dominant familial exudative vitreoretinopathy. Br. J. Ophthalmol. 2015, 100, 278–283. [Google Scholar] [CrossRef] [Green Version]
- Li, V.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.; Mohammed, S.; Heck, A.; Maurice, M.; Mahmoudi, T.; et al. Wnt Signaling through Inhibition of β-Catenin Degradation in an Intact Axin1 Complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Zhang, X.; Zhang, Q.; Zhao, P. Variable reduction in Norrin signaling activity caused by novel mutations in FZD4 identified in patients with familial exudative vitreoretinopathy. Mol. Vis. 2019, 25, 60–69. [Google Scholar] [PubMed]
- Pendergast, S.D.; Trese, M.T. Familial exudative vitreoretinopathy. Results of surgical management. Ophthalmology 1998, 105, 1015–1023. [Google Scholar] [CrossRef]
- Ranchod, T.M.; Ho, L.Y.; Drenser, K.A.; Capone, A.; Trese, M.T. Clinical Presentation of Familial Exudative Vitreoretinopathy. Ophthalmology 2011, 118, 2070–2075. [Google Scholar] [CrossRef]
- Ohlmann, A.; Scholz, M.; Goldwich, A.; Chauhan, B.K.; Hudl, K.; Ohlmann, A.V.; Zrenner, E.; Berger, W.; Cvekl, A.; Seeliger, M.W.; et al. Ectopic Norrin Induces Growth of Ocular Capillaries and Restores Normal Retinal Angiogenesis in Norrie Disease Mutant Mice. J. Neurosci. 2005, 25, 1701–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karjosukarso, D.W.; Van Gestel, S.H.C.; Qu, J.; Kouwenhoven, E.N.; Duijkers, L.; Garanto, A.; Zhou, H.; Collin, R.W.J. An FEVR-associated mutation in ZNF408 alters the expression of genes involved in the development of vasculature. Hum. Mol. Genet. 2018, 27, 3519–3527. [Google Scholar] [CrossRef]
- Li, J.-K.; Li, Y.; Zhang, X.; Chen, C.-L.; Rao, Y.-Q.; Fei, P.; Zhang, Q.; Zhao, P.; Li, J. Spectrum of Variants in 389 Chinese Probands With Familial Exudative Vitreoretinopathy. Investig. Opthalmol. Vis. Sci. 2018, 59, 5368–5381. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Tong, Y.; Zhu, Y.; Peng, M. Familial Exudative Vitreoretinopathy-Related Disease-Causing Genes and Norrin/ β -Catenin Signal Pathway: Structure, Function, and Mutation Spectrums. J. Ophthalmol. 2019, 2019, 5782536. [Google Scholar] [CrossRef]
- Seo, S.H.; Yu, Y.S.; Park, S.W.; Kim, J.H.; Kim, H.K.; Cho, S.I.; Park, H.; Lee, S.J.; Seong, M.-W.; Park, S.S.; et al. Molecular Characterization of FZD4, LRP5, and TSPAN12 in Familial Exudative Vitreoretinopathy. Investig. Opthalmol. Vis. Sci. 2015, 56, 5143. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.L.; Parry, N.R.; Barton, S.J.; Campbell, C.; Delaney, C.M.; Ellingford, J.; Hall, G.; Hardcastle, C.; Morarji, J.; Nichol, E.J.; et al. Panel-Based Clinical Genetic Testing in 85 Children with Inherited Retinal Disease. Ophthalmology 2017, 124, 985–991. [Google Scholar] [CrossRef]
- LaDuca, H.; Farwell, K.D.; Vuong, H.; Lu, H.-M.; Mu, W.; Shahmirzadi, L.; Tang, S.; Chen, J.; Bhide, S.; Chao, E.C. Exome sequencing covers >98% of mutations identified on targeted next generation sequencing panels. PLoS ONE 2017, 12, e0170843. [Google Scholar] [CrossRef] [PubMed]
- Consugar, M.B.; Navarro-Gomez, D.; Place, E.M.; Bujakowska, K.M.; Sousa, M.E.; Fonseca-Kelly, Z.D.; Taub, D.G.; Janessian, M.; Wang, D.Y.; Au, E.D.; et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 2014, 17, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Number of amplicon regions | 180.0 |
| Total length of target regions | 32,731.0 |
| Percent on-target aligned reads | 98.3 |
| Percent on-target PF reads | 92.9 |
| Percent aligned read | 94.6 |
| Percent on-target aligned bases | 98.4 |
| Percent on-target PF bases | 92.2 |
| Percent Q30 bases | 95.5 |
| Percent aligned bases | 93.7 |
| Amplicon mean coverage | 978.0 |
| Variant Type | Total Count | Average per Sample |
|---|---|---|
| SNVs total | 937 | 19.5 |
| SNVs in genes | 927 | 19.3 |
| SNVs in exons | 268 | 5.6 |
| SNVs in coding regions | 220 | 4.6 |
| SNVs in UTR regions | 48 | 1.0 |
| SNVs in splice site regions | 55 | 1.1 |
| Stop-gained SNVs | 2 | 0.04 |
| Stop-lost SNVs | 0 | 0.0 |
| Non-synonymous SNVs | 44 | 0.9 |
| Synonymous SNVs | 174 | 3.6 |
| SNVs not in dbSNP | 3 | 0.1 |
| Insertions total | 81 | 1.7 |
| Insertions in genes | 28 | 0.6 |
| Insertions in exons | 10 | 0.2 |
| Insertions in coding regions | 8 | 0.2 |
| Insertions in UTR regions | 2 | 0.04 |
| Insertions in splice site regions | 0 | 0.0 |
| Stop-gained insertions | 0 | 0.0 |
| Stop-lost insertions | 0 | 0.0 |
| Frame shift insertions | 2 | 0.04 |
| Non-synonymous insertions | 6 | 0.1 |
| Insertions not in dbSNP | 3 | 0.1 |
| Deletions total | 71 | 1.5 |
| Deletions in genes | 26 | 0.5 |
| Deletions in exons | 19 | 0.4 |
| Deletions in coding regions | 15 | 0.3 |
| Deletions in UTR regions | 4 | 0.1 |
| Deletions in splice site regions | 0 | 0.0 |
| Stop-gained deletions | 0 | 0.0 |
| Stop-lost deletions | 0 | 0.0 |
| Frame shift deletions | 0 | 0.0 |
| Non-synonymous deletions | 15 | 0.3 |
| Deletions not in dbSNP | 10 | 0.2 |
| Group | FEVR Grade | Family | Gender | Age | Relationship | Coding-Variations | AF 1 | AF 2 | AF 3 | AF 4 | Combined AF | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | NA | NA | 1 | M | 43 | maternal uncle of 4 and 5 | LRP5:Val 667Met LRP5:Ala1330Val | 0.034 | 0.134 | NA | NA | 0.004556 |
| 2 | NA | NA | 1 | F | 44 | maternal aunt of 4 and 5 | LRP5:Val 667Met LRP5:Ala1330Val | 0.034 | 0.134 | NA | NA | 0.004556 |
| 3 | NA | NA | 1 | F | 41 | mother of 4 and 5 | LRP5:Val 667Met LRP5:Ala1330Val | 0.034 | 0.134 | NA | NA | 0.004556 |
| 4 | FEVR | 5 | 1 | M | 1 | pediatric, son, proband 1 | LRP5:Val 667Met LRP5:Ala1330Val (homo) | 0.034 | 0.016 | NA | NA | 0.000544 |
| 5 | FEVR | 4 | 1 | M | 1 | pediatric, twin of 4 | LRP5:Val 667Met LRP5:Ala1330Val | 0.034 | 0.134 | NA | NA | 0.004556 |
| 6 | FEVR | 1 | 2 | F | 42 | mother of 7 | LRP5: Leu20dup ZNF408:Arg337Pro | 0.1014 | 0.0086 | NA | NA | 0.000872 |
| 7 | FEVR | 2 | 2 | M | 1 | pediatric, son, proband 2 | CTTNB1:Val273Met | 0.000021 | NA | NA | NA | 0.000021 |
| 8 | NA | NA | 3 | M | 67 | father of 9 | LRP5:Ala1330Val | 0.134 | NA | NA | NA | 0.134 |
| 9 | FEVR | 4 | 3 | M | 29 | son, proband 3 | LRP5:Leu20dup LRP5:Ala1330Val (homo) | 0.1014 | 0.016 | NA | NA | 0.001622 |
| 10 | U | 0 | 4 | F | 28 | mother of pediatric not collected yet | LRP5: Met1086Val ZNF408:Arg337Pro | 0.002077 | 0.0086 | NA | NA | 0.000018 |
| 11 | U | 0 | 4 | M | 30 | father of pediatric not collected yet | No protein changes | NA | NA | NA | NA | NA |
| 12 | U | 0 | 5 | F | 42 | mother of 14 | LRP5:Leu20dup ZNF408:Val194_Val197del | 0.1014 | 0.1307 | NA | NA | 0.013253 |
| 13 | FEVR | 1 | 5 | M | 54 | father of 14 | LRP5:Val667Met LRP5:Ala1330Val ZNF408:Val194_Val197del | 0.034 | 0.134 | 0.1307 | NA | 0.000595 |
| 14 | FEVR | 3 | 5 | F | 3 | pediatric, daughter, proband-5 | LRP5:Leu20dup ZNF408:Val194_Val197del | 0.1014 | 0.1307 | NA | NA | 0.013253 |
| 15 | NA | NA | 7 | M | 45 | father of 17 and 18 | FZD4:Met105Val LRP5:Ala1330Val ZNF408:Val194_Val197del | 0.000043 | 0.134 | 0.1307 | NA | 0.000001 |
| 16 | NA | NA | 7 | F | 43 | mother of 17 | No protein changes | NA | NA | NA | NA | NA |
| 17 | FEVR | 2 | 7 | F | 16 | pediatric, daughter, proband-7 | FZD4:Met105Val LRP5:Ala1330Val | 0.000043 | 0.134 | NA | NA | 0.000006 |
| 18 | NA | NA | 7 | F | 19 | sibling of 17 | ZNF408:Val194_Val197del | 0.1307 | NA | NA | NA | 0.1307 |
| 19 | FEVR | 1 | 8 | F | 14 | pediatric, daughter, proband-8 | KIF11:Glu129Ala LRP5:Ala1330Val (homo) | 0 | 0.016 | NA | NA | 0 |
| 20 | U | 0 | 8 | F | 11 | pediatric, sibling of 19 | ZNF408:Val194_Val197del | 0.1307 | NA | NA | NA | 0.1307 |
| 21 | U | 0 | 8 | M | 13 | pediatric, sibling of 19 | LRP5:Ala1330Val | 0.134 | NA | NA | NA | 0.134 |
| 22 | U | 0 | 9 | M | 31 | father of 23 | No protein changes | NA | NA | NA | NA | NA |
| 23 | FEVR | 5 | 9 | M | 1 | pediatric, son, proband-9 | FZD4:Met105Val | 0.000043 | NA | NA | NA | 0.000043 |
| 24 | FEVR | 1 | 10 | M | 57 | maternal uncle of 26 | LRP5:c.4488+2T>G (splice donor) | 0 | NA | NA | NA | 0 |
| 25 | FEVR | 1 | 10 | F | 45 | mother of 26 | LRP5:c.4488+2T>G (splice donor) LRP5:Leu20dup | 0 | 0.1014 | NA | NA | 0 |
| 26 | FEVR | 5 | 10 | F | 18 | daughter, proband-10 | LRP5:c.4488+2T>G (splice donor) FZD4:Pro11Gln | 0 | 0.000021 | NA | NA | 0 |
| 27 | U | 0 | 10 | M | 17 | son of maternal uncle 24 | No protein changes | NA | NA | NA | NA | NA |
| 28 | U | 0 | 11 | M | 68 | maternal grandfather of 30 | LRP5:Leu20dup ZNF408:Val194_Val197del | 0.1014 | 0.1307 | NA | NA | 0.013253 |
| 29 | FEVR | 1 | 11 | F | 30 | mother of 30 | FZD4:Cys450ter LRP5:Leu20dup | 0 | 0.1014 | NA | NA | 0 |
| 30 | FEVR | 5 | 11 | F | 3 | pediatric, daughter, proband-11 | FZD4:Cys450ter FZD4:Met105Val | 0 | 0.000043 | NA | NA | 0 |
| 31 | NA | NA | 12 | M | 28 | father of 33 | LRP5:Val667Met LRP5:Ala1330Val ZNF408:Val194_Val197del | 0.034 | 0.134 | 0.1307 | NA | 0.000595 |
| 32 | NA | NA | 12 | F | 26 | mother of 33 | No protein changes | NA | NA | NA | NA | NA |
| 33 | FEVR | 5 | 12 | F | 1 | pediatric, daughter, proband-12 | LRP5:Val667Met LRP5:Ala1330Val | 0.034 | 0.134 | NA | 0.004556 | |
| 34 | NA | NA | 13 | F | 74 | grandmother of 38 | No protein changes | NA | NA | NA | NA | NA |
| 35 | FEVR | 1 | 13 | M | 73 | grandfather of 38 | LRP5:Val667Met LRP5:Ala1330Val FZD4:Pro33Ser FZD4:Pro168Ser | 0.034 | 0.134 | 0.01236 | 0.014077 | 0.000001 |
| 36 | U | 0 | 13 | M | 49 | father of 39 | LRP5:Val667Met LRP5:Ala1330Val ZNF408:Val194_Val197del | 0.034 | 0.134 | 0.1307 | NA | 0.000595 |
| 37 | FEVR | 1 | 13 | F | 47 | mother of 38 | No protein changes | NA | NA | NA | NA | NA |
| 38 | FEVR | 4 | 13 | M | 11 | pediatric, son, proband-13 | LRP5:Val667Met LRP5:Ala1330Val | 0.034 | 0.134 | NA | NA | 0.004556 |
| 39 | U | 0 | 13 | F | 12 | pediatric, daughter, sibling of 38 | LRP5:Val667Met LRP5:Ala1330Val | 0.034 | 0.134 | NA | NA | 0.004556 |
| 40 | FEVR | 1 | 14 | F | 46 | mother of 41 | LRP5:Leu16_Leu20del ZNF408:Val194_Val197del | 0.000007 | 0.1307 | NA | NA | 0.000001 |
| 41 | FEVR | 4 | 14 | M | 6 | pediatric, son, proband-14 | LRP5:c.4488+2T>G (splice donor) | 0 | NA | NA | NA | 0 |
| 42 | FEVR | 2 | 15 | M | 3 | pediatric, son, twin of 43 | ZNF408:Glu230Gly | 0.000371 | NA | NA | NA | 0.000371 |
| 43 | FEVR | 1 | 15 | M | 3 | pediatric, son, twin of 42 | ZNF408:Glu230Gly | 0.000371 | NA | NA | NA | 0.000371 |
| 44 | NA | NA | 16 | F | 8 | pediatric, daughter, sibling of 47 | LRP5:Ala1330Val ZNF408:Val194_Val197del | 0.134 | 0.1307 | NA | NA | 0.017514 |
| 45 | U | 0 | 16 | M | 11 | sibling of 46 | LRP5: Ala1330Val ZNF408:Val194_Val197del | 0.134 | 0.1307 | NA | NA | 0.017514 |
| 46 | NA | NA | 17 | M | NA | father of child (FEVR not sequenced) | ZNF408:Val194_Val197del | 0.1307 | NA | NA | NA | 0.1307 |
| 47 | NA | NA | 17 | F | 37 | mother of child (FEVR not sequenced) | ZNF408:Val194_Val197del | 0.1307 | NA | NA | NA | 0.1307 |
| 48 | NA | NA | 18 | M | 41 | father of child (FEVR not sequenced) | FZD4:Arg127Cys LRP5:Val667Met LRP5:Ala1330Val | 0.000046 | 0.034 | 0.134 | NA | 0 |
| 49 | U | 0 | 18 | F | 38 | mother of child (FEVR not sequenced) | No protein changes | NA | NA | NA | NA | 0 |
| 50 | NA | NA | 19 | F | 60 | mother of child (FEVR not sequenced) | LRP5:Pro6Thr ZNF408:Val194_Val197del | 0.002932 | 0.1307 | NA | NA | 0.000383 |
| 51 | FEVR | 3 | NA | F | 2 | pediatric, no relations | LRP5:Val667Met LRP5:Ala1330Val LRP5:Pro848Leu LRP5:Thr852Met | 0.034 | 0.134 | 0 | 0.000013 | 0 |
| 52 | FEVR | 1 | NA | F | 90 | adult, no relations | KIF11:His526Gln ZNF408:Val194_Val197del | 0.002532 | 0.1307 | NA | NA | 0.000331 |
| 53 | FEVR | 2 | NA | M | 11 | pediatric, no relations | LRP5:Ala1330Val ZNF408:Val194_Val197del(homo) | 0.134 | 0.0159 | NA | NA | 0.002131 |
| 54 | FEVR | 4 | NA | F | 6 | pediatric, no relations | LRP5:Leu20dup ZNF408:Val194_Val197del | 0.1014 | 0.1307 | NA | NA | 0.013253 |
| 55 | FEVR | 1 | NA | F | 54 | adult, no relations | LRP5:Ala1330Val | 0.134 | NA | NA | NA | 0.134 |
| 56 | FEVR | 4 | NA | M | 9 | pediatric, no relations | NDP:His42Arg | 0.000005 | NA | NA | NA | 0.000005 |
| 57 | FEVR | 2 | NA | M | 1 | pediatric, no relations | LRP5:Val667Met LRP5:Ala1330Val ZNF408:Val194_Val197del | 0.034 | 0.134 | 0.1307 | NA | 0.000595 |
| 58 | FEVR | 5 | NA | F | 1 | pediatric, no relations | LRP5:Cys913LeufsTer73 | 0.000004 | NA | NA | NA | 0.000004 |
| 59 | FEVR | 5 | NA | F | 1 | pediatric, no relations | LRP5:Ala919CysfsTer67 | 0 | NA | NA | NA | 0 |
| 60 | FEVR | 1 | NA | M | 14 | pediatric, no relations | LRP5:Leu20dup | 0.1014 | NA | NA | NA | 0.1014 |
| 61 | FEVR | 5 | NA | M | 1 | pediatric, no relations | LRP5:Pro1522Leu FZD4:Ala408del | 0.000291 | 0 | NA | NA | 0 |
| 62 | FEVR | 3 | NA | F | 34 | adult, no relations | LRP5:Val667Met LRP5:Ala1330Val ZNF408:Val194_Val197del FZD4 Gly161Arg | 0.034 | 0.134 | 0.1307 | 0 | 0 |
| 63 | FROP | 2 | NA | M | 2 | pediatric, no relations | No protein changes | NA | NA | NA | NA | NA |
| 64 | FROP | NA | NA | F | 1 | pediatric, no relations | ZNF408:Val194_Val197del | 0.1307 | NA | NA | NA | 0.1307 |
| 65 | FROP | 5 | NA | F | 1 | pediatric, no relations | LRP5:Gln89Arg | 0.008262 | NA | NA | NA | 0.008262 |
| 66 | FROP | 5 | NA | F | 1 | pediatric, no relations | No protein changes | NA | NA | NA | NA | NA |
| 67 | FROP | 4 | NA | F | 39 | adult, no relations | LRP5:Ala1330Val | 0.134 | NA | NA | NA | 0.134 |
| 68 | U | 0 | NA | M | 1 | pediatric, no relations | LRP5:Leu20dup | 0.1014 | NA | NA | NA | 0.1014 |
| 69 | U | 0 | NA | M | 7 | pediatric, no relations | ZNF408:Val194_Val197del | 0.1307 | NA | NA | NA | 0.1307 |
| 70 | ROP | NA | NA | F | 1 | pediatric, no relations | ZNF408:Val194_Val197del FZD4:Trp139Ser | 0.1307 | 0 | NA | NA | 0 |
| 71 | U | 0 | NA | F | 9 | adult, no relations | LRP5:Leu20dup | 0.1014 | NA | NA | NA | 0.1014 |
| 72 | U | 0 | NA | F | 15 | pediatric, no relations | No protein changes | NA | NA | NA | NA | NA |
| 73 | U | 0 | NA | M | 14 | pediatric, no relations | KIF11:Pro642Ala | 0.000293 | NA | NA | NA | 0.000293 |
| 74 | U | 0 | NA | F | 69 | adult, no relations | LRP5:Leu20dup | 0.1014 | NA | NA | NA | 0.1014 |
| 75 | U | 0 | NA | M | 43 | adult, no relations | No protein changes | NA | NA | NA | NA | NA |
| 76 | U | 0 | NA | F | 6 | pediatric, no relations | ZNF408:Leu67Val ZNF408:Ala372Thr ZNF408:Pro647Gln | 0.00368 | 0.000577 | 0 | NA | 0 |
| No. | Gene | Nucleotide | Protein | Molecular Change | Consequence | AF % | AF (Homo) % | dbSNP | ClinVar Accession |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CTNNB1 | NM_001098209.2:c.817G>A | NP_001091679.1:p.Val273Met | missense variant | (Uncertain significance) | 0.0021% | 0.0% | rs1183899293 | Not in ClinVar |
| 2 | FZD4 | NM_012193.4:c.32C>A | NP_036325.2:p.Pro11Gln | missense variant | (Uncertain significance) | 0.0021% | 0.0% | rs766393047 | Not in ClinVar |
| 3 | FZD4 | NM_012193.4:c.97C>T | NP_036325.2:p.Pro33Ser | missense variant | Benign | 1.236% | 0.89% | rs61735304 | RCV000387944.3 |
| 4 | FZD4 | NM_012193.4:c.313A>G | NP_036325.2:p.Met105Val | missense variant | Pathogenic | 0.0043% | 0.0% | rs80358284 | RCV000210241.1 |
| 5 | FZD4 | NM_012193.4:c.379C>T | NP_036325.2:p.Arg127Cys | missense variant | Likely benign | 0.0046% | 0.0% | rs376854255 | RCV001111581.1 |
| 6 New | FZD4 | NM_012193.4:c.416G>C | NP_036325.2:p.Trp139Ser | missense variant | (Uncertain significance) | NA | NA | NA | Not in ClinVar |
| 7 New | FZD4 | NM_012193.4:c.481G>C | NP_036325.2:p.Gly161Arg | missense variant | (Uncertain significance) | NA | NA | NA | Not in ClinVar |
| 8 | FZD4 | NM_012193.4:c.502C>T | NP_036325.2:p.Pro168Ser | missense variant | Benign | 1.4077% | 0.01379% | rs61735303 | RCV000368489.2 |
| 9 New | FZD4 | NM_012193.4:c.1221_1223delTCG | NP_036325.2:p.Ala408del | in-frame deletion | (Uncertain significance) | NA | NA | NA | Not in ClinVar |
| 10 New | FZD4 | NM_012193.4:c.1350T>A | NP_036325.2:p.Cys450Ter | stop-gained | (Likely pathogenic) | NA | NA | NA | Not in ClinVar |
| 11 | KIF11 | NM_004523.4:c.386A>C | NP_004514.2:p.Glu129Ala | missense variant | (Uncertain significance) | 0.00004 | NA | rs779558239 | Not in ClinVar |
| 12 | KIF11 | NM_004523.4:c.1924C>G | NP_004514.2:p.Pro642Ala | missense variant | Benign | 0.02930% | 0.0% | rs79865214 | RCV000915477.2 |
| 13 | KIF11 | NM_004523.4:c.1578C>A | NP_004514.2:p.His526Gln | missense variant | Benign | 0.25320% | 0.0013% | rs112145870 | RCV000967853.2 |
| 14 | LRP5 | NM_002335.4:c.16C>A | NP_002326.2:p.Pro6Thr | missense variant | Benign | 0.2932% | 0.0035% | rs771718186 | RCV000592927.1 |
| 15 | LRP5 | NM_002335.4:c.34_36CTG [4] | NP_002326.2:p.Leu16_Leu20del | deletion | Uncertain significance | 0.0006928% | 0.0% | rs72555376 | Not in ClinVar |
| 16 | LRP5 | NM_002335.4:c.58_60dupCTG | NP_002326.2:p.Leu20dup | insert duplication | Benign | 10.14% | 0.57% | rs564221347 | VCV000193231.2 |
| 17 | LRP5 | NM_002335.4:c.266A>G | NP_002326.2:p.Gln89Arg | missense variant | Benign | 0.83% | NA | rs41494349 | RCV000175719.2 |
| 18 | LRP5 | NM_002335.4:c.1999G>A | NP_002326.2:p.Val667Met | missense variant | Likely benign | 3.40% | 0.092% | rs4988321 | RCV000250939.1 |
| 19 New | LRP5 | NM_002335.4:c.2543C>T | NP_002326.2:p.Pro848Leu | missense variant | NA | NA | NA | NA | Not in ClinVar |
| 20 | LRP5 | NM_002335.4:c.2555C>T | NP_002326.2:p.Thr852Met | missense variant | NA | 0.0013140% | 0.0% | rs1398692057 | Not in ClinVar |
| 21 | LRP5 | NM_002335.4:c.2737dup | NP_002326.2:p.Cys913fsTer73 | frameshift variant | Pathogenic | 0.0004000% | 0.0% | rs886043590 | RCV000761295.1 |
| 22 New | LRP5 | NM_002335.4:c.2754dup | NP_002326.2:p.Ala919CysfsTer67 | frameshift variant | (Likely pathogenic) | NA | NA | NA | Not in ClinVar |
| 23 | LRP5 | NM_002335.4:c.3256A>G | NP_002326.2:p.Met1086Val | missense variant | Likely benign | 0.20770% | 0.0019730% | rs145774832 | RCV000592263.1 |
| 24 | LRP5 | NM_002335.4:c.3989C>T | NP_002326.2:p.Ala1330Val | missense variant | Benign | 13.40% | 1.60% | rs3736228 | RCV000242123.2 |
| 25 | LPR5 | NM_002335.4:c.4565C>T | NP_002326.2:p.Pro1522Leu | missense variant | Uncertain significance | 0.02910% | 0.0% | rs200624778 | RCV000724481.4 |
| 26 | LRP5 | NM_002335.4:c.4488+2T>G | intron 21 donor site lost | splice donor | Pathogenic | NA | NA | rs80358322 | RCV000006667.3 |
| 27 | NDP | NM_000266.4:c.125A>G | NP_000257.1:p.His42Arg | missense variant | Pathogenic | 0.0005% | NA | rs104894874 | RCV000011437.5 |
| 28 | ZNF408 | NM_024741.3:c.199C>G | NP_079017.1:p.Leu67Val | missense variant | Benign | 0.3680% | 0.0019710% | rs35652367 | RCV000969067.2 |
| 29 | ZNF408 | NM_024741.3:c.581_592del | NP_079017.1:p.Val194_Val197del | deletion | Benign | 13.1% | 1.59% | rs148055528 | RCV000248955.1 |
| 30 | ZNF408 | NM_024741.3:c.689A>G | NP_079017.1:p.Glu230Gly | missense variant | Likely benign | 0.0371% | 0.0006570% | rs147850078 | RCV000974525.2 |
| 31 | ZNF408 | NM_024741.3:c.1010G>C | NP_079017.1:p.Arg337Pro | missense variant | Benign | 0.8609% | 0.0151100% | rs36017347 | RCV000959911.2 |
| 32 | ZNF408 | NM_024741.3:c.1114G>A | NP_079017.1:p.Ala372Thr | missense variant | Uncertain significance | 0.0577% | 0.0% | rs141624151 | RCV001053827.1 |
| 33 New | ZNF408 | NM_024741.3:c.1940_1941delinsAA | NP_079017.1:p.Pro647Gln | missense variant | (Uncertain significance) | NA | NA | NA | Not in ClinVar |
| Variant Nature | Gene/s | Total |
|---|---|---|
| monogenic | NDP | 1 |
| monogenic | LRP5 | 16 |
| monogenic | LRP5 (2) | 7 |
| monogenic | LRP5 (3) | 1 |
| monogenic | FZD4 | 2 |
| monogenic | FZD4 (2) | 1 |
| monogenic | CTNNB1 | 1 |
| monogenic | ZNF408 | 1 |
| monogenic | ZNF408 (3) | 1 |
| monogenic | KIFll | 1 |
| digenic | LRP5, ZNF408 | 14 |
| digenic | LRP5, FZD4 | 6 |
| digenic | LRP5, KIF11 | 2 |
| digenic | KIF11, ZNF408 | 1 |
| digenic | FZD4, ZNF408 | 1 |
| trigenic | LRP5, ZNF408, FZD4 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicerone, A.P.; Dailey, W.; Sun, M.; Santos, A.; Jeong, D.; Jones, L.; Koustas, K.; Drekh, M.; Schmitz, K.; Haque, N.; et al. A Survey of Multigenic Protein-Altering Variant Frequency in Familial Exudative Vitreo-Retinopathy (FEVR) Patients by Targeted Sequencing of Seven FEVR-Linked Genes. Genes 2022, 13, 495. https://doi.org/10.3390/genes13030495
Cicerone AP, Dailey W, Sun M, Santos A, Jeong D, Jones L, Koustas K, Drekh M, Schmitz K, Haque N, et al. A Survey of Multigenic Protein-Altering Variant Frequency in Familial Exudative Vitreo-Retinopathy (FEVR) Patients by Targeted Sequencing of Seven FEVR-Linked Genes. Genes. 2022; 13(3):495. https://doi.org/10.3390/genes13030495
Chicago/Turabian StyleCicerone, Amanda Petrelli, Wendy Dailey, Michael Sun, Andrew Santos, Daeun Jeong, Lance Jones, Konstaninos Koustas, Mary Drekh, Keaton Schmitz, Naomi Haque, and et al. 2022. "A Survey of Multigenic Protein-Altering Variant Frequency in Familial Exudative Vitreo-Retinopathy (FEVR) Patients by Targeted Sequencing of Seven FEVR-Linked Genes" Genes 13, no. 3: 495. https://doi.org/10.3390/genes13030495
APA StyleCicerone, A. P., Dailey, W., Sun, M., Santos, A., Jeong, D., Jones, L., Koustas, K., Drekh, M., Schmitz, K., Haque, N., Felisky, J. A., Guzman, A. E., Mellert, K., Trese, M. T., Capone, A., Drenser, K. A., & Mitton, K. P. (2022). A Survey of Multigenic Protein-Altering Variant Frequency in Familial Exudative Vitreo-Retinopathy (FEVR) Patients by Targeted Sequencing of Seven FEVR-Linked Genes. Genes, 13(3), 495. https://doi.org/10.3390/genes13030495