
Deconstructing Immune Cell Infiltration in Human Colorectal Cancer: A Systematic Spatiotemporal Evaluation

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Immunohistochemical Staining of CRC Samples
2.2. NanoString Pan-Cancer Immune Panel
2.3. Description of Clinicopathological Data
2.4. Statistical Analysis
3. Results
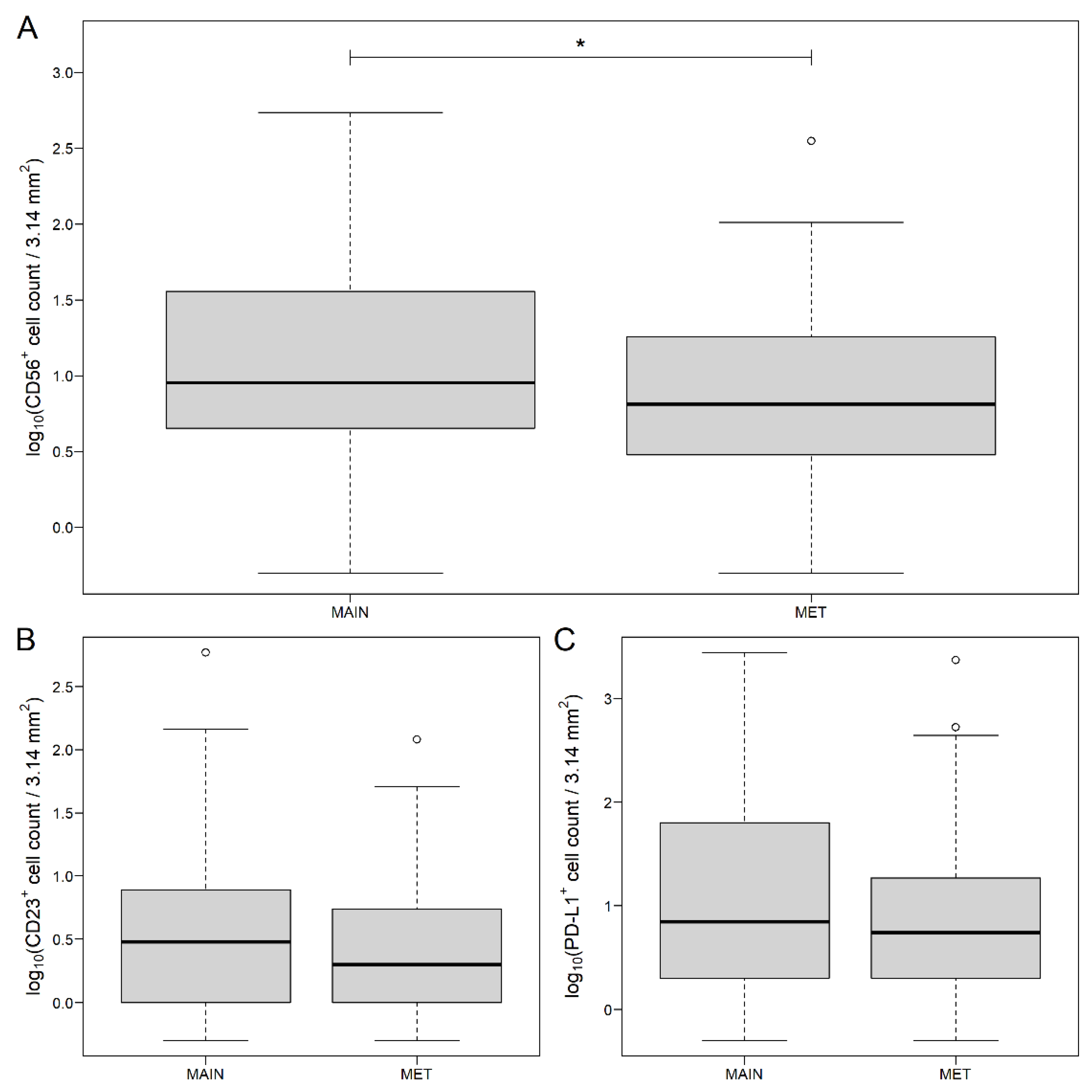
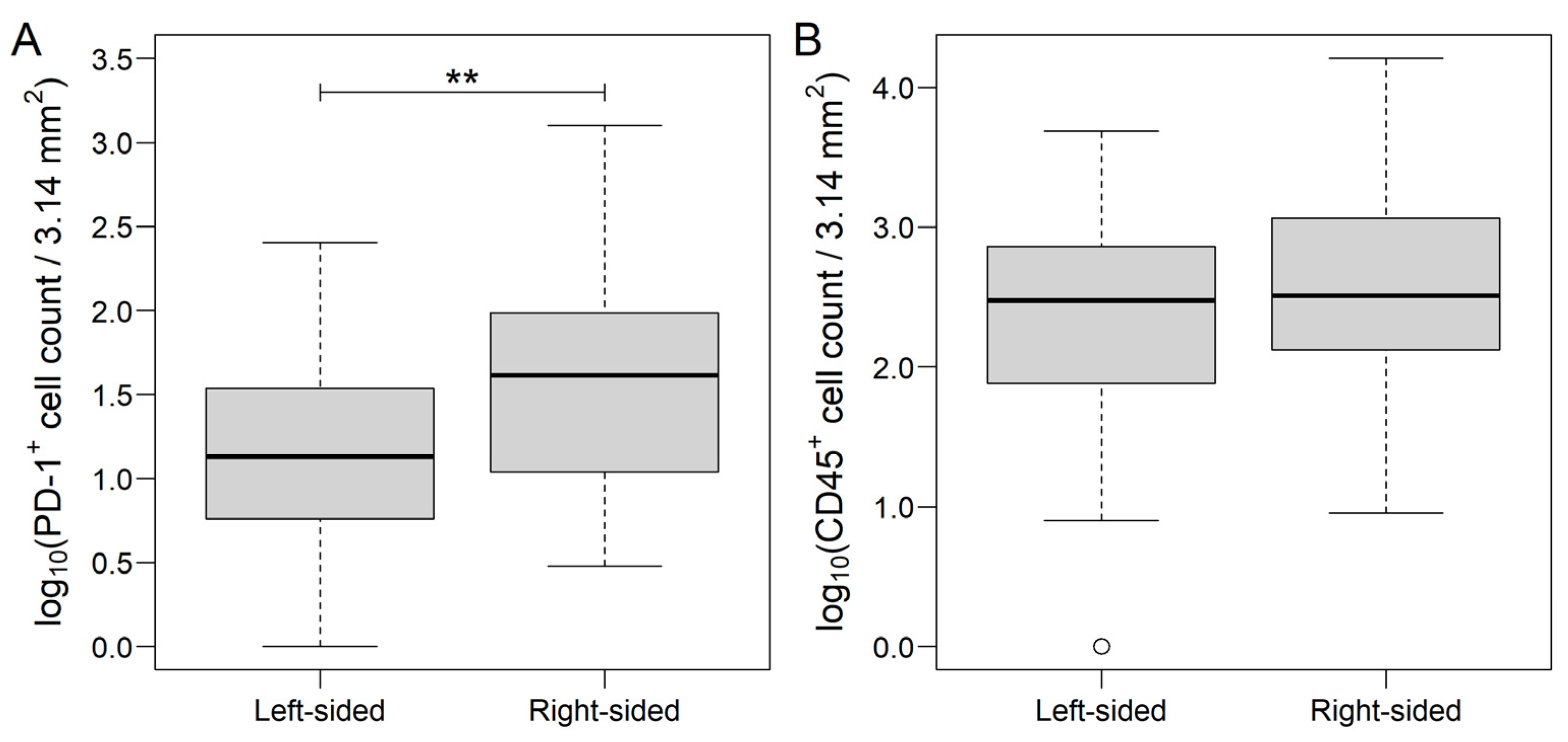
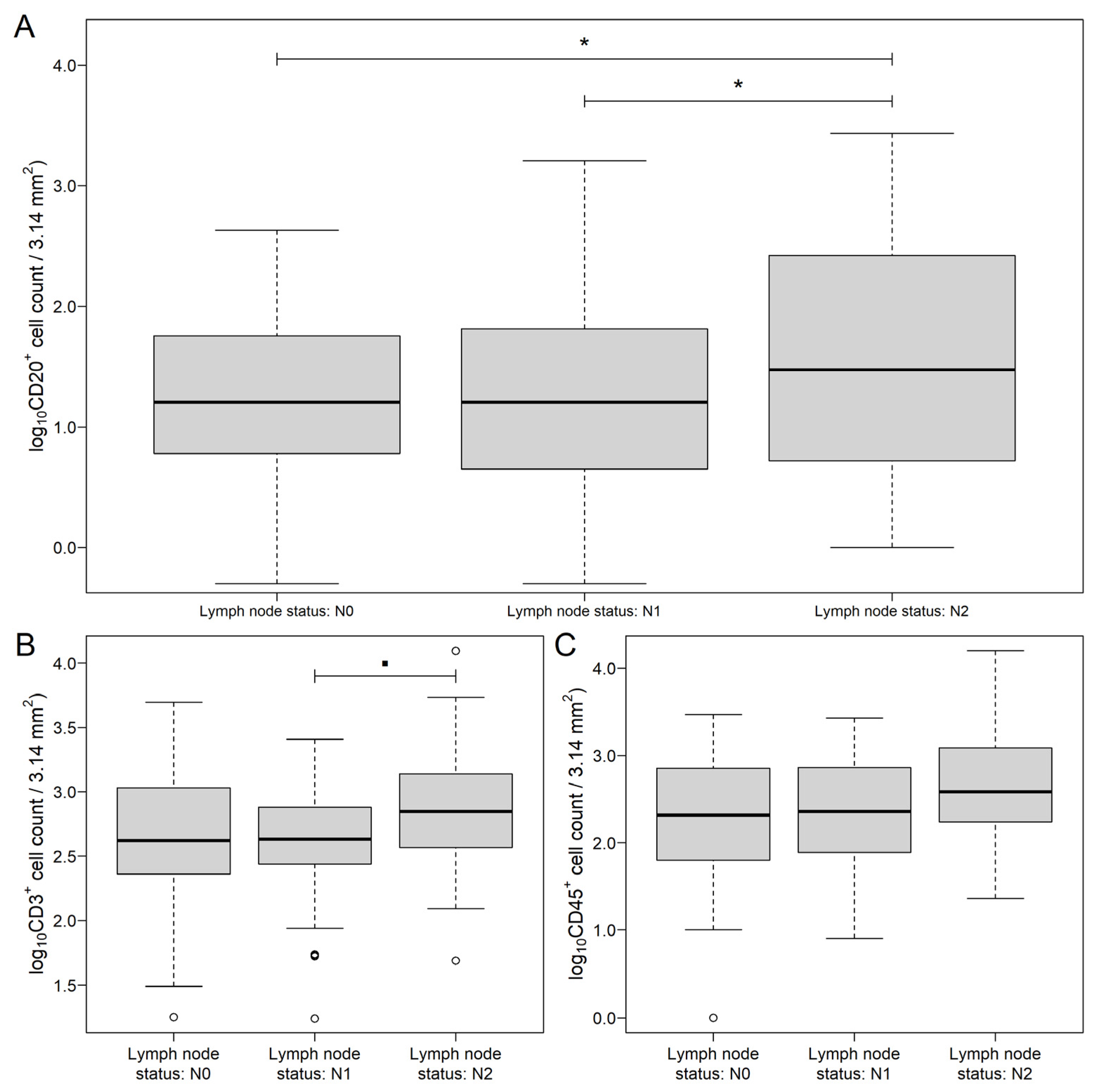
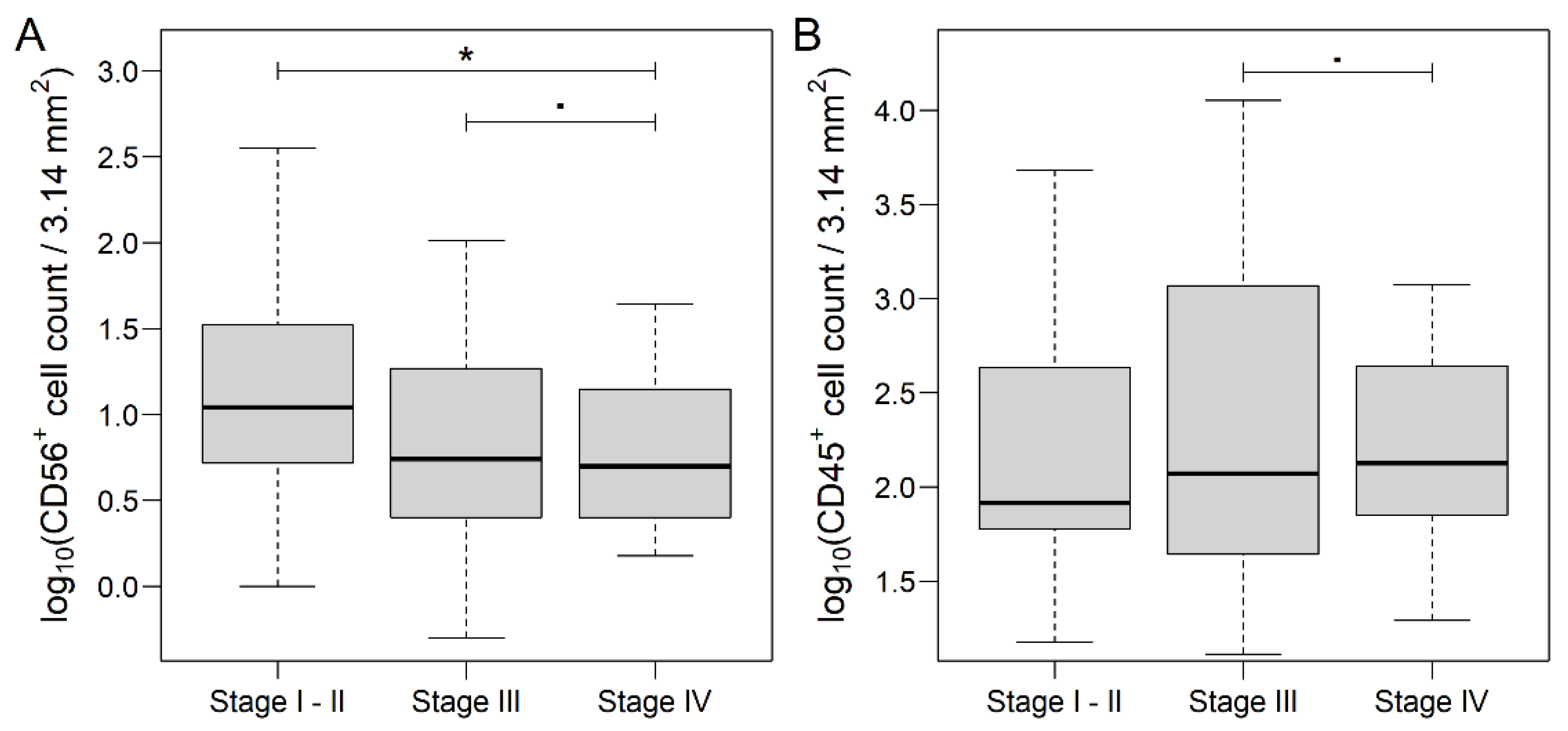
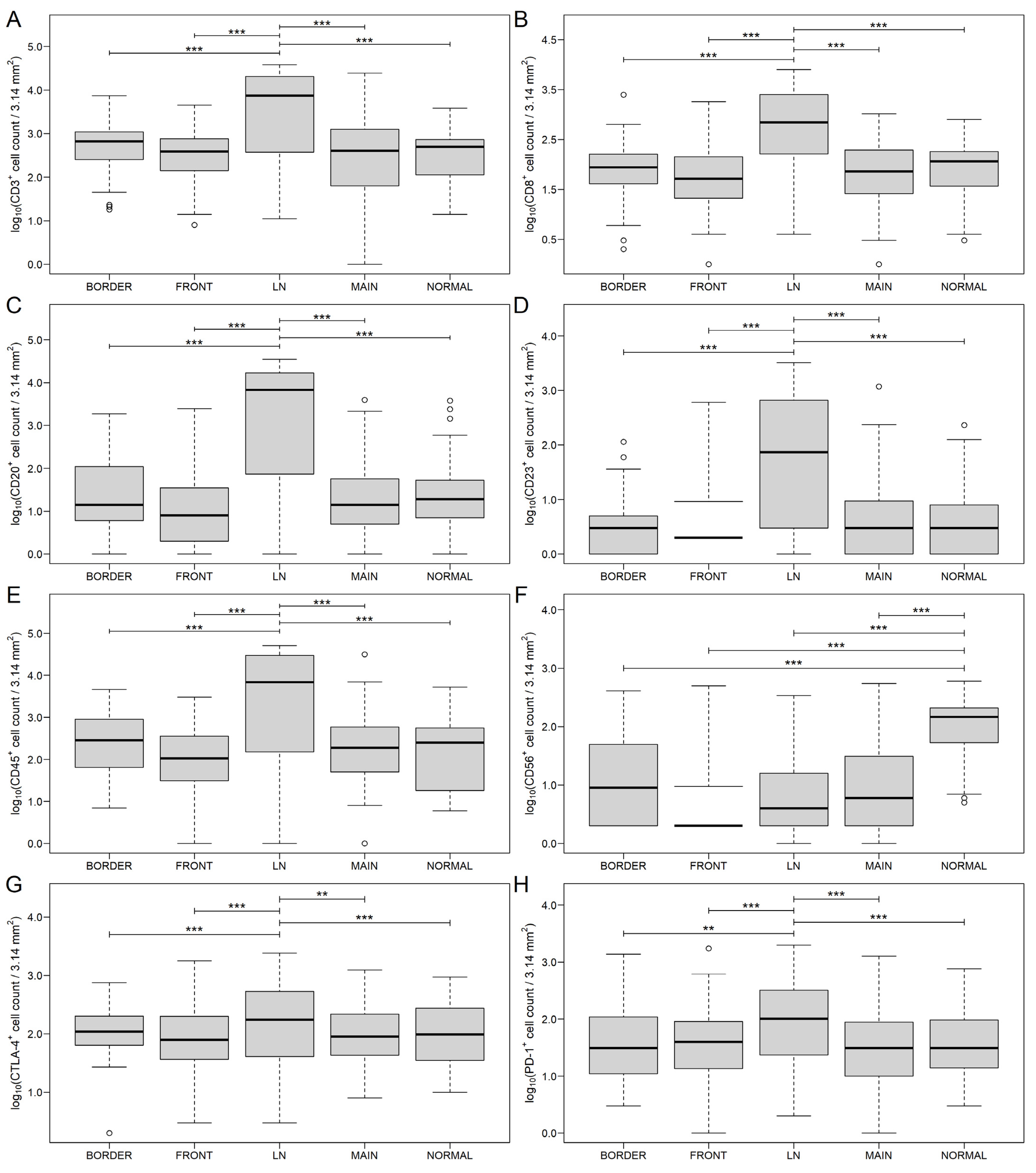
3.1. Distribution of Markers in Different Areas of CRC and Metastatic Lymph Nodes
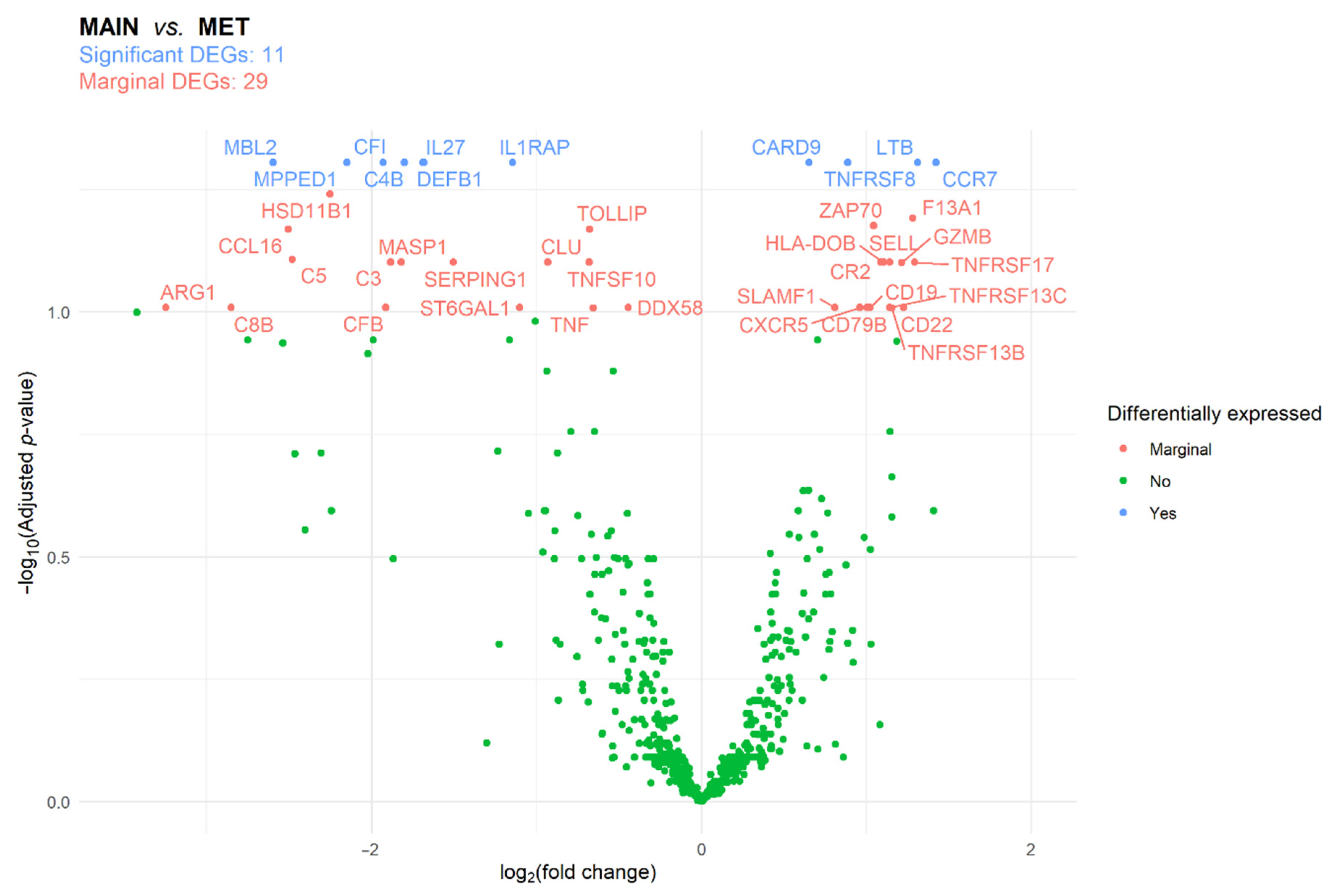
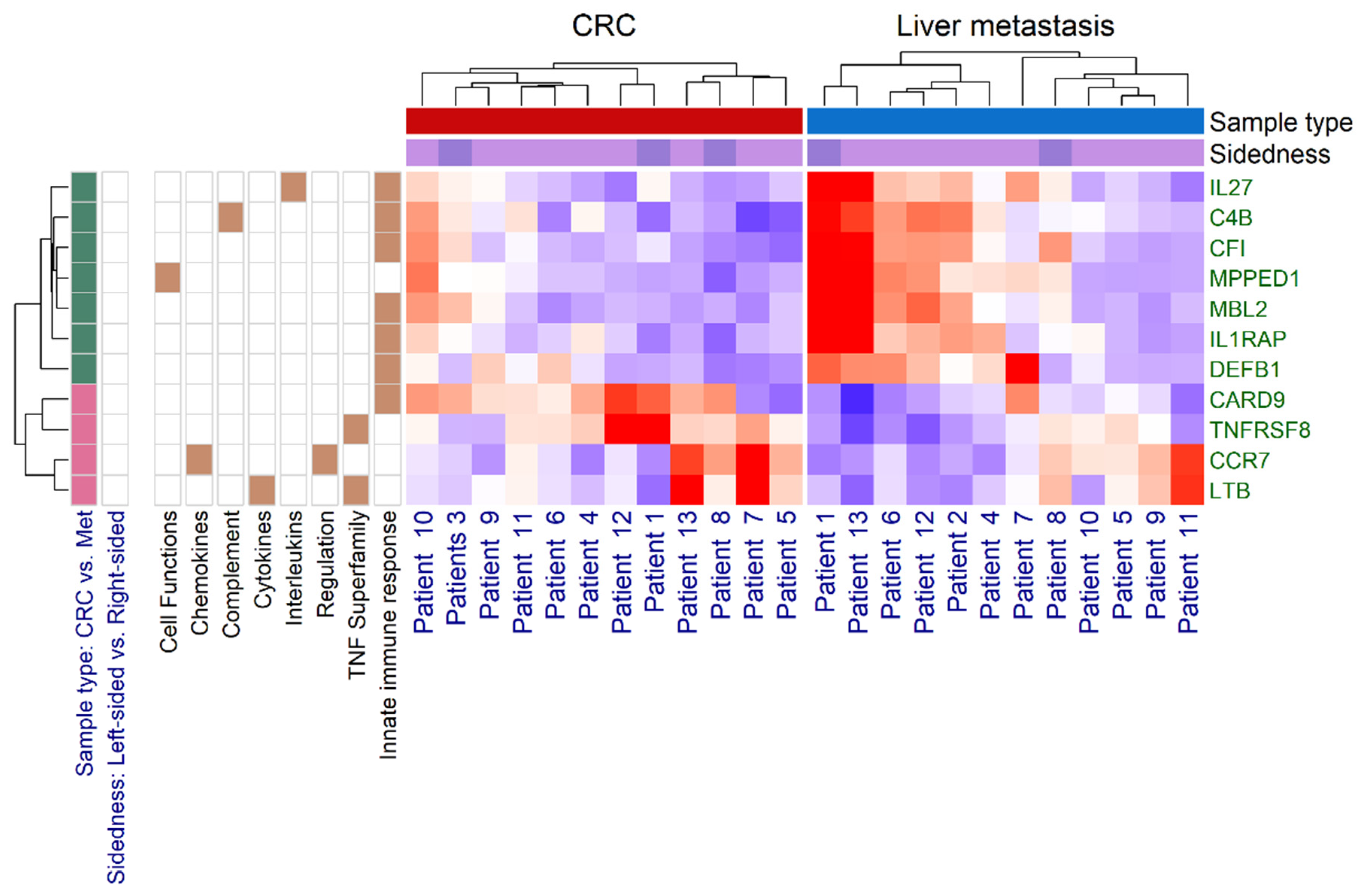
3.2. Immune Panel Gene Expression Analysis
4. Discussion
Limitations of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Roxburgh, C.S.; McMillan, D.C. The role of the in situ local inflammatory response in predicting recurrence and survival in patients with primary operable colorectal cancer. Cancer Treat Rev. 2012, 38, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.H.; Pages, F.; et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Funada, Y.; Noguchi, T.; Kikuchi, R.; Takeno, S.; Uchida, Y.; Gabbert, H.E. Prognostic significance of CD8+ T cell and macrophage peritumoral infiltration in colorectal cancer. Oncol. Rep. 2003, 10, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Freeman, G.J. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015, 5, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Jessup, J.; Goldberg, R.; Asare, E.; Benson, A.; Brierley, J.; Chang, G.; Chen, V.; Compton, C.; De Nardi, P.; Goodman, K.; et al. Colon and Rectum. In AJCC Cancer Staging Manual, 8th ed.; Amin, M., Edge, S., Greene, F., Byrd, D., Brookland, R., Washington, M., Gershenwald, J., Compton, C., Hess, K., Sullivan, D., et al., Eds.; Springer International Publishing: Chicago, IL, USA, 2018; pp. 251–274. [Google Scholar]
- Shen, H.; Yang, J.; Huang, Q.; Jiang, M.J.; Tan, Y.N.; Fu, J.F.; Zhu, L.Z.; Fang, X.F.; Yuan, Y. Different treatment strategies and molecular features between right-sided and left-sided colon cancers. World J. Gastroenterol. 2015, 21, 6470–6478. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Hamilton, A.M.; Furberg, H.; Pietzak, E.; Purdue, M.P.; Troester, M.A.; Hoadley, K.A.; Love, M.I. An approach for normalization and quality control for NanoString RNA expression data. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis, 3rd ed.; Springer: New York, NY, USA, 2016. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienstmann, R.; Salazar, R.; Tabernero, J. Molecular Subtypes and the Evolution of Treatment Decisions in Metastatic Colorectal Cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 231–238. [Google Scholar] [CrossRef]
- Taniguchi, H.; Nakamura, Y.; Kotani, D.; Yukami, H.; Mishima, S.; Sawada, K.; Shirasu, H.; Ebi, H.; Yamanaka, T.; Aleshin, A.; et al. CIRCULATE-Japan: Circulating tumor DNA-guided adaptive platform trials to refine adjuvant therapy for colorectal cancer. Cancer Sci. 2021, 112, 2915–2920. [Google Scholar] [CrossRef]
- Becht, E.; de Reynies, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautes-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [Green Version]
- Kather, J.N.; Halama, N.; Jaeger, D. Genomics and emerging biomarkers for immunotherapy of colorectal cancer. Semin. Cancer Biol. 2018, 52, 189–197. [Google Scholar] [CrossRef]
- Van den Eynde, M.; Mlecnik, B.; Bindea, G.; Fredriksen, T.; Church, S.E.; Lafontaine, L.; Haicheur, N.; Marliot, F.; Angelova, M.; Vasaturo, A.; et al. The Link between the Multiverse of Immune Microenvironments in Metastases and the Survival of Colorectal Cancer Patients. Cancer Cell 2018, 34, 1012–1026.e1013. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Wang, C.; Qiu, X.; Pu, X.; Chang, P. Colorectal Cancer Immune Infiltrates: Significance in Patient Prognosis and Immunotherapeutic Efficacy. Front. Immunol. 2020, 11, 1052. [Google Scholar] [CrossRef]
- Kurashima, Y.; Kiyono, H. Mucosal Ecological Network of Epithelium and Immune Cells for Gut Homeostasis and Tissue Healing. Annu. Rev. Immunol. 2017, 35, 119–147. [Google Scholar] [CrossRef]
- Zirakzadeh, A.A.; Marits, P.; Sherif, A.; Winqvist, O. Multiplex B cell characterization in blood, lymph nodes, and tumors from patients with malignancies. J. Immunol. 2013, 190, 5847–5855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazama, K.; Otake, J.; Satoyoshi, T.; Shiozawa, M.; Sugano, N.; Sato, S.; Atsumi, Y.; Kano, K.; Murakawa, M.; Maezawa, Y.; et al. Distribution of Regulatory T-Cells and Other Phenotypes of T-Cells in Tumors and Regional Lymph Nodes of Colorectal Cancer Patients. In Vivo 2020, 34, 849–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betts, G.; Jones, E.; Junaid, S.; El-Shanawany, T.; Scurr, M.; Mizen, P.; Kumar, M.; Jones, S.; Rees, B.; Williams, G.; et al. Suppression of tumour-specific CD4(+) T cells by regulatory T cells is associated with progression of human colorectal cancer. Gut 2012, 61, 1163–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chruscik, A.; Gopalan, V.; Lam, A.K. The clinical and biological roles of transforming growth factor β in colon cancer stem cells: A systematic review. Eur. J. Cell Biol. 2018, 97, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Pugh, S.A.; Harrison, R.J.; Primrose, J.N.; Khakoo, S.I. T cells but not NK cells are associated with a favourable outcome for resected colorectal liver metastases. BMC Cancer 2014, 14, 180. [Google Scholar] [CrossRef] [Green Version]
- Le Voyer, T.E.; Sigurdson, E.R.; Hanlon, A.L.; Mayer, R.J.; Macdonald, J.S.; Catalano, P.J.; Haller, D.G. Colon cancer survival is associated with increasing number of lymph nodes analyzed: A secondary survey of intergroup trial INT-0089. J. Clin. Oncol. 2003, 21, 2912–2919. [Google Scholar] [CrossRef]
- Kim, Y.W.; Jan, K.M.; Jung, D.H.; Cho, M.Y.; Kim, N.K. Histological inflammatory cell infiltration is associated with the number of lymph nodes retrieved in colorectal cancer. Anticancer Res. 2013, 33, 5143–5150. [Google Scholar]
- George, S.; Primrose, J.; Talbot, R.; Smith, J.; Mullee, M.; Bailey, D.; du Boulay, C.; Jordan, H.; Wessex Colorectal Cancer Audit Working Group. Will Rogers revisited: Prospective observational study of survival of 3592 patients with colorectal cancer according to number of nodes examined by pathologists. Br. J. Cancer 2006, 95, 841–847. [Google Scholar] [CrossRef] [Green Version]
- Samaržija, I. Wnt Signaling Pathway Is among the Drivers of Liver Metastasis. Livers 2021, 1, 180–200. [Google Scholar] [CrossRef]
- Liu, J.; Wang, D.; Zhang, C.; Zhang, Z.; Chen, X.; Lian, J.; Liu, J.; Wang, G.; Yuan, W.; Sun, Z.; et al. Identification of liver metastasis-associated genes in human colon carcinoma by mRNA profiling. Chin. J. Cancer Res. 2018, 30, 633–646. [Google Scholar] [CrossRef]
- Nadal, C.; Maurel, J.; Gascon, P. Is there a genetic signature for liver metastasis in colorectal cancer? World J. Gastroenterol. 2007, 13, 5832–5844. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, F.; Jiang, Q.; Sun, Y.; Liao, Q.; An, H.; Li, Y.; Li, Z.; Fan, L.; Guo, F.; et al. Gene Expression Profiling for Differential Diagnosis of Liver Metastases: A Multicenter, Retrospective Cohort Study. Front. Oncol. 2021, 11, 725988. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; McCormick, T.S.; Weinberg, A. Human β Defensins and Cancer: Contradictions and Common Ground. Front. Oncol. 2019, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Talaat, I.M.; Elemam, N.M.; Saber-Ayad, M. Complement System: An Immunotherapy Target in Colorectal Cancer. Front. Immunol. 2022, 13, 810993. [Google Scholar] [CrossRef]
- Tian, W.; Jiang, X.; Kim, D.; Guan, T.; Nicolls, M.R.; Rockson, S.G. Leukotrienes in Tumor-Associated Inflammation. Front. Pharmacol. 2020, 11, 1289. [Google Scholar] [CrossRef]
- Hartjes, L.; Ruland, J. CARD9 Signaling in Intestinal Immune Homeostasis and Oncogenesis. Front. Immunol. 2019, 10, 419. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Number of Observation/Mean ± SD |
|---|---|
| Age (year) | 60.58 ± 11.01 |
| Sex (Male:Female) | 77:60 (56.2%:43.8%) |
| Location of the CRC 1 | |
| – Coecum | 16 (11.7%) |
| – Ascending colon | 16 (11.7%) |
| – Transverse colon | 11 (8%) |
| – Descending colon | 15 (10.9%) |
| – Sigmoid colon | 50 (36.5%) |
| – Rectum | 28 (20.4%) |
| Sidedness of the tumor | |
| – Right-sided | 45 (32.8%) |
| – Left-sided | 92 (67.2%) |
| pT—extent of the tumor 1 | |
| – T1 | 1 (0.7%) |
| – T2 | 11 (8%) |
| – T3 | 93 (67.9%) |
| – T4 | 27 (19.7%) |
| pN—lymph node status 1 | |
| – N0 | 44 (32.1%) |
| – N1 | 48 (35%) |
| – N2 | 42 (30.7%) |
| AJCC [9] staging 1 | |
| – Stage I | 2 (1.5%) |
| – Stage II | 27 (19.7%) |
| – Stage III | 54 (39.4%) |
| – Stage IV | 51 (37.2%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ágoston, E.I.; Acs, B.; Herold, Z.; Fekete, K.; Kulka, J.; Nagy, A.; Mühl, D.; Mohacsi, R.; Dank, M.; Garay, T.; et al. Deconstructing Immune Cell Infiltration in Human Colorectal Cancer: A Systematic Spatiotemporal Evaluation. Genes 2022, 13, 589. https://doi.org/10.3390/genes13040589
Ágoston EI, Acs B, Herold Z, Fekete K, Kulka J, Nagy A, Mühl D, Mohacsi R, Dank M, Garay T, et al. Deconstructing Immune Cell Infiltration in Human Colorectal Cancer: A Systematic Spatiotemporal Evaluation. Genes. 2022; 13(4):589. https://doi.org/10.3390/genes13040589
Chicago/Turabian StyleÁgoston, Emese Irma, Balazs Acs, Zoltan Herold, Krisztina Fekete, Janina Kulka, Akos Nagy, Dorottya Mühl, Reka Mohacsi, Magdolna Dank, Tamas Garay, and et al. 2022. "Deconstructing Immune Cell Infiltration in Human Colorectal Cancer: A Systematic Spatiotemporal Evaluation" Genes 13, no. 4: 589. https://doi.org/10.3390/genes13040589
APA StyleÁgoston, E. I., Acs, B., Herold, Z., Fekete, K., Kulka, J., Nagy, A., Mühl, D., Mohacsi, R., Dank, M., Garay, T., Harsanyi, L., Győrffy, B., & Szasz, A. M. (2022). Deconstructing Immune Cell Infiltration in Human Colorectal Cancer: A Systematic Spatiotemporal Evaluation. Genes, 13(4), 589. https://doi.org/10.3390/genes13040589