
Rare and Common Variants Uncover the Role of the Atria in Coarctation of the Aorta

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
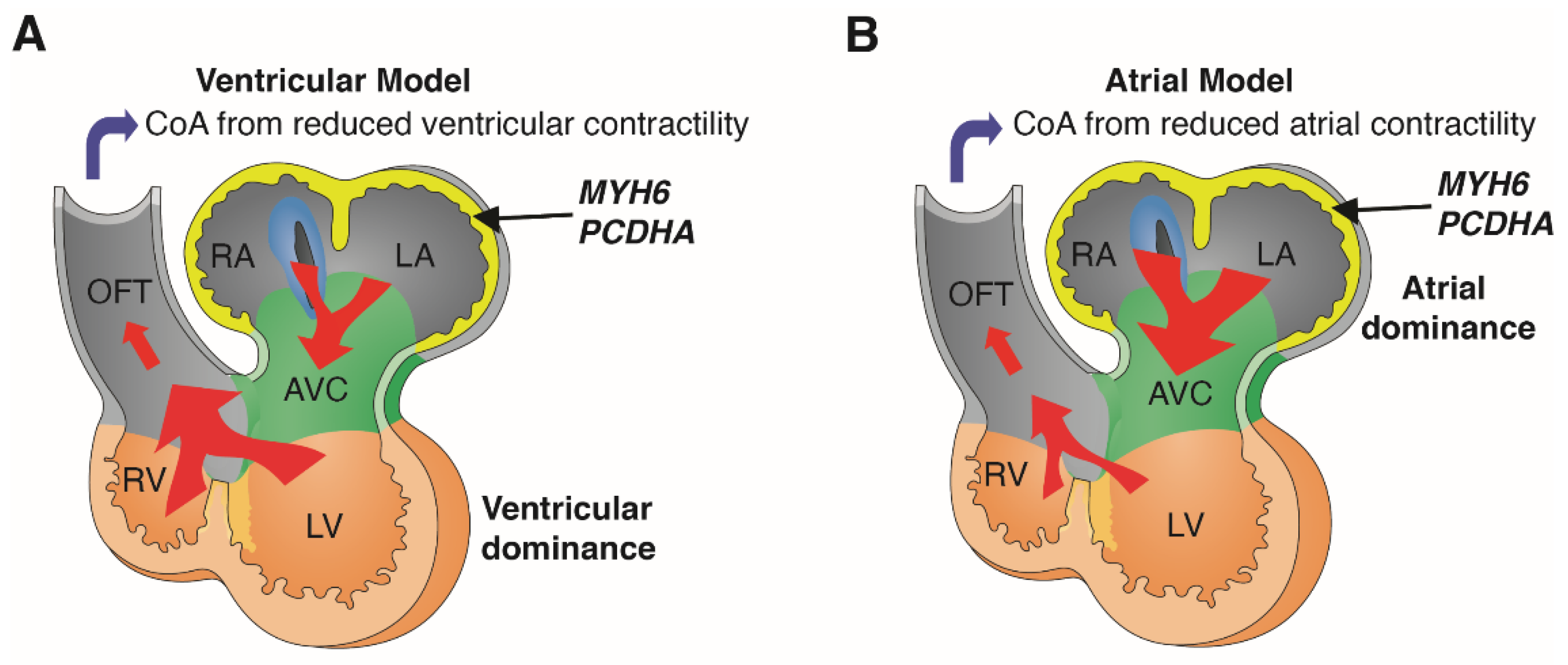
:1. Introduction
2. Materials and Methods
2.1. Human Study Participants
2.2. Recovery of Rare Predicted Pathogenic MYH6 Variants
2.3. Gene-Based Burden Testing
2.4. Identification of TagSNPs for the 16.8 kb PCDHA delCNV in European-American Population
2.5. Determining eQTL Tissue Association Using Genotype-Tissue Expression (GTEx) Database
2.6. Analysis of Publicly Available Single Cell RNA Sequencing Data
2.7. Additional Information on Data Presented in This Study
3. Statistics
4. Results
4.1. Significant Association of Rare MYH6 Variants with CoA and BAV
4.2. Cooccurence of Rare MYH6 Variants with the PCDHA delCNV
4.3. MYH6 and PCDHA Expression in the Human and Mouse Heart
4.4. eQTL Shows PCHDA delCNV Associated with Reduced PCDHA10 Expression in Human Atria
5. Discussion
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCDHA | The protocadherin α cluster |
| PCDHA10 | Protocadherin α 10 |
| MYH6 | Cardiac α (α)-myosin heavy chain |
| CoA | Coarctation of aorta |
| BAV | Bicuspid aortic valve |
| HLHS | Hypoplastic left heart syndrome |
| LVOTO | Left ventricular outflow tract obstructions |
| eQTLs | Expression quantitative trait loci |
| PCDHA delCNVs | Deletion copy number variants in PCDHA |
| GnomAD | The Genome Aggregation Database |
References
- Granados-Riveron, J.T.; Brook, J.D. The impact of mechanical forces in heart morphogenesis. Circ. Cardiovasc. Genet. 2012, 5, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Midgett, M.; Thornburg, K.; Rugonyi, S. Blood flow patterns underlie developmental heart defects. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H632–H642. [Google Scholar] [CrossRef] [PubMed]
- Steed, E.; Boselli, F.; Vermot, J. Hemodynamics driven cardiac valve morphogenesis. Biochim. Biophys. Acta 2016, 1863, 1760–1766. [Google Scholar] [CrossRef]
- Hoog, T.G.; Fredrickson, S.J.; Hsu, C.W.; Senger, S.M.; Dickinson, M.E.; Udan, R.S. The effects of reduced hemodynamic loading on morphogenesis of the mouse embryonic heart. Dev. Biol. 2018, 442, 127–137. [Google Scholar] [CrossRef]
- Baumgartner, H.; Bonhoeffer, P.; De Groot, N.M.; de Haan, F.; Deanfield, J.E.; Galie, N.; Gatzoulis, M.A.; Gohlke-Baerwolf, C.; Kaemmerer, H.; Kilner, P.; et al. ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur. Heart J. 2010, 31, 2915–2957. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, A.M.; Heymann, M.A.; Spitznas, U. Hemodynamic considerations in the development of narrowing of the aorta. Am. J. Cardiol. 1972, 30, 514–525. [Google Scholar] [CrossRef]
- Allen, D.D.; Shaddy, R.E.; Feltes, T.F. Moss & Adams’ Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adult, 8th ed.; Lippincott Wilkins & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Siu, S.C.; Silversides, C.K. Bicuspid aortic valve disease. J. Am. Coll. Cardiol. 2010, 55, 2789–2800. [Google Scholar] [CrossRef] [Green Version]
- Parker, L.E.; Landstrom, A.P. Genetic Etiology of Left-Sided Obstructive Heart Lesions: A Story in Development. J. Am. Heart Assoc. 2021, 10, e019006. [Google Scholar] [CrossRef]
- Sanchez-Cascos, A. The recurrence risk in congenital heart disease. Eur. J. Cardiol. 1978, 7, 197–210. [Google Scholar]
- Bjornsson, T.; Thorolfsdottir, R.B.; Sveinbjornsson, G.; Sulem, P.; Norddahl, G.L.; Helgadottir, A.; Gretarsdottir, S.; Magnusdottir, A.; Danielsen, R.; Sigurdsson, E.L.; et al. A rare missense mutation in MYH6 associates with non-syndromic coarctation of the aorta. Eur. Heart J. 2018, 39, 3243–3249. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Fleres, B.; Lovett, J.; Anfinson, M.; Samudrala, S.S.K.; Kelly, L.J.; Teigen, L.E.; Cavanaugh, M.; Marquez, M.; Geurts, A.M.; et al. Contractility of Induced Pluripotent Stem Cell-Cardiomyocytes With an MYH6 Head Domain Variant Associated With Hypoplastic Left Heart Syndrome. Front. Cell Dev. Biol. 2020, 8, 440. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Zhu, W.; Gabriel, G.C.; Young, C.B.; Williams, K.; Martin, L.J.; Hill, J.C.; Richards, T.; Billaud, M.; Phillippi, J.A.; et al. Common Deletion Variants Causing Protocadherin-α Deficiency Contribute to the Complex Genetics of Bicuspid Aortic Valve and Left-sided Congenital Heart Disease. Hum. Genet. Genom. Adv. 2021, 2, 100037. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef]
- Guo, M.H.; Plummer, L.; Chan, Y.M.; Hirschhorn, J.N.; Lippincott, M.F. Burden Testing of Rare Variants Identified through Exome Sequencing via Publicly Available Control Data. Am. J. Hum. Genet. 2018, 103, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Mazzarotto, F.; Tayal, U.; Buchan, R.J.; Midwinter, W.; Wilk, A.; Whiffin, N.; Govind, R.; Mazaika, E.; de Marvao, A.; Dawes, T.J.W.; et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020, 141, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Xu, A.; Sim, S.; Priest, J.R.; Tian, X.; Khan, T.; Quertermous, T.; Zhou, B.; Tsao, P.S.; Quake, S.R.; et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev. Cell 2016, 39, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Chhibbar, P.; Lo, C. The transcriptional landscape of the clustered protocadherins in the cardiovascular system. Eur. Heart J. 2021, 42, 3202. [Google Scholar] [CrossRef]
- Reiser, P.J.; Portman, M.A.; Ning, X.H.; Schomisch Moravec, C. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1814-1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Furth, D.; Qian, X.; Wardell, E.; Custodio, J.; Reimegard, J.; Salmen, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e1619. [Google Scholar] [CrossRef]
- Noguchi, Y.; Hirabayashi, T.; Katori, S.; Kawamura, Y.; Sanbo, M.; Hirabayashi, M.; Kiyonari, H.; Nakao, K.; Uchimura, A.; Yagi, T. Total expression and dual gene-regulatory mechanisms maintained in deletions and duplications of the Pcdha cluster. J. Biol. Chem. 2009, 284, 32002–32014. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yagi, H.; Saeed, S.; Bais, A.S.; Gabriel, G.C.; Chen, Z.; Peterson, K.A.; Li, Y.; Schwartz, M.C.; Reynolds, W.T.; et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017, 49, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- van Eif, V.W.W.; Devalla, H.D.; Boink, G.J.J.; Christoffels, V.M. Transcriptional regulation of the cardiac conduction system. Nat. Rev. Cardiol. 2018, 15, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Wloch, A.; Rozmus-Warcholinska, W.; Cnota, W.; Huhta, J.; Acharya, G. Atrial dominance in the human embryonic heart: A study of cardiac function at 6–10 weeks of gestation. Ultrasound Obstet. Gynecol. 2015, 46, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Sheikh, F.; Hollander, M.; Cai, C.; Becker, D.; Chu, P.H.; Evans, S.; Chen, J. Embryonic atrial function is essential for mouse embryogenesis, cardiac morphogenesis and angiogenesis. Development 2003, 130, 6111–6119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, A.; Agnese, V.; Coronnello, C.; Raffa, G.M.; Bellavia, D.; Conaldi, P.G.; Pilato, M.; Pasta, S. On the prospect of serum exosomal miRNA profiling and protein biomarkers for the diagnosis of ascending aortic dilatation in patients with bicuspid and tricuspid aortic valve. Int. J. Cardiol. 2018, 273, 230–236. [Google Scholar] [CrossRef]
- Pasta, S.; Agnese, V.; Gallo, A.; Cosentino, F.; Di Giuseppe, M.; Gentile, G.; Raffa, G.M.; Maalouf, J.F.; Michelena, H.I.; Bellavia, D.; et al. Shear Stress and Aortic Strain Associations With Biomarkers of Ascending Thoracic Aortic Aneurysm. Ann. Thorac. Surg. 2020, 110, 1595–1604. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Williams, K.; Young, C.; Lin, J.-H.; Teekakirikul, P.; Lo, C.W. Rare and Common Variants Uncover the Role of the Atria in Coarctation of the Aorta. Genes 2022, 13, 636. https://doi.org/10.3390/genes13040636
Zhu W, Williams K, Young C, Lin J-H, Teekakirikul P, Lo CW. Rare and Common Variants Uncover the Role of the Atria in Coarctation of the Aorta. Genes. 2022; 13(4):636. https://doi.org/10.3390/genes13040636
Chicago/Turabian StyleZhu, Wenjuan, Kylia Williams, Cullen Young, Jiaunn-Huey Lin, Polakit Teekakirikul, and Cecilia W. Lo. 2022. "Rare and Common Variants Uncover the Role of the Atria in Coarctation of the Aorta" Genes 13, no. 4: 636. https://doi.org/10.3390/genes13040636
APA StyleZhu, W., Williams, K., Young, C., Lin, J. -H., Teekakirikul, P., & Lo, C. W. (2022). Rare and Common Variants Uncover the Role of the Atria in Coarctation of the Aorta. Genes, 13(4), 636. https://doi.org/10.3390/genes13040636