
Pcsk6 Deficiency Promotes Cardiomyocyte Senescence by Modulating Ddit3-Mediated ER Stress

 , and
, and
Abstract
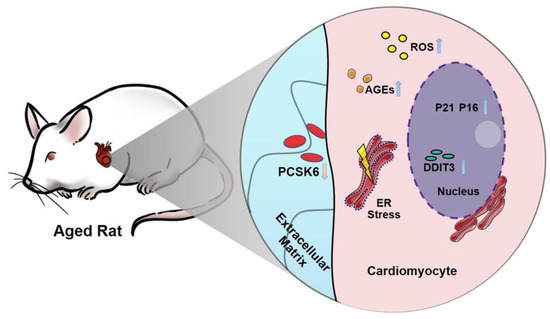
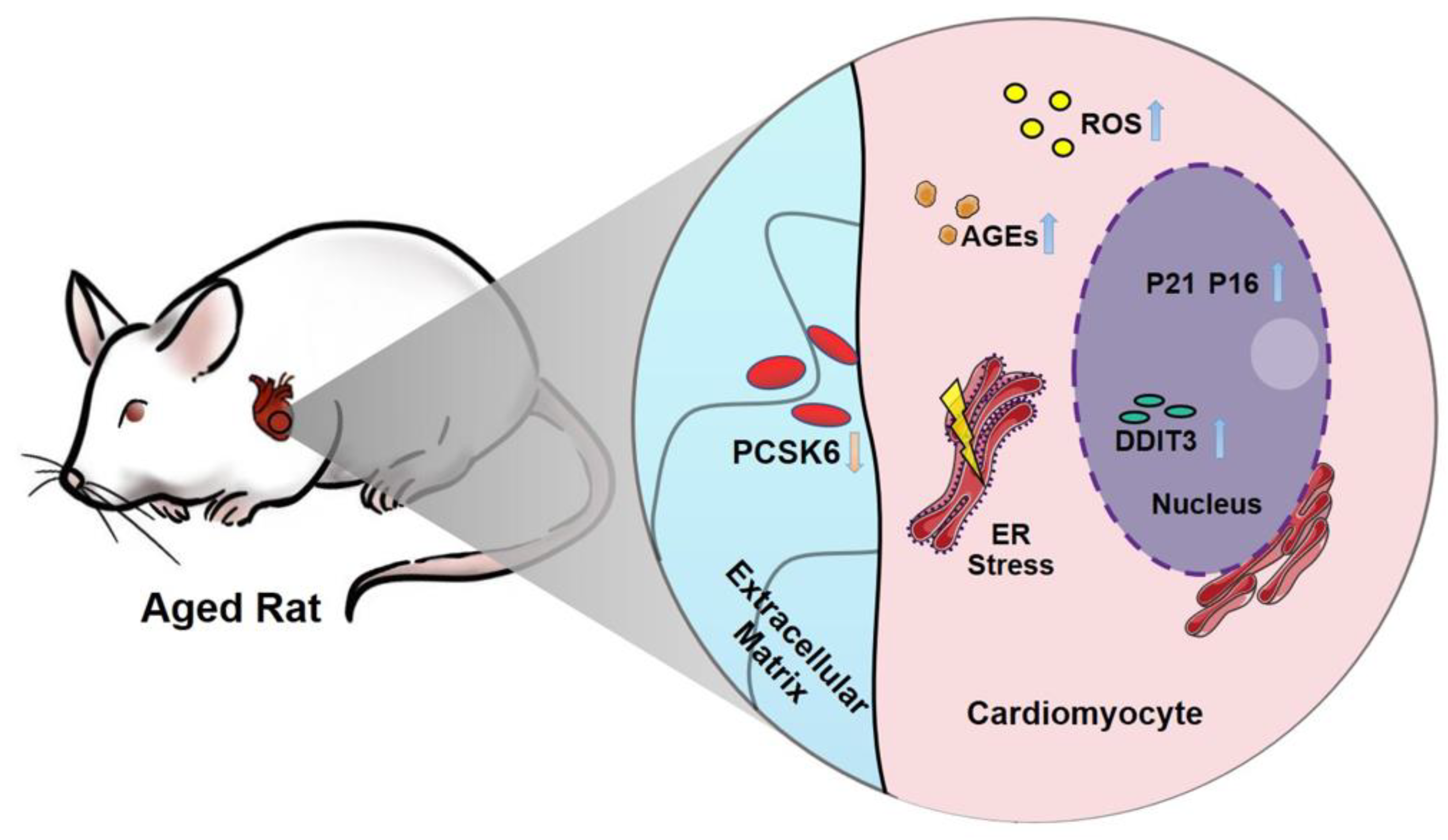
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Cell Culture and Treatment
2.3. SiRNA and Plasmid DNA Transfection
2.4. RNA Sequencing and Analysis
2.5. Immunofluorescent Staining
2.6. Hoechst Staining
2.7. Senescence-Associated β-Galactosidase (SA-β-Gal) Staining
2.8. RNA Isolation and Quantitative Real-Time PCR (qPCR)
2.9. Protein Extraction and Western Blotting
2.10. Measurement of Reactive Oxygen Species (ROS)
2.11. Measurement of Apoptotic Activity
2.12. Statistical Analysis
3. Results
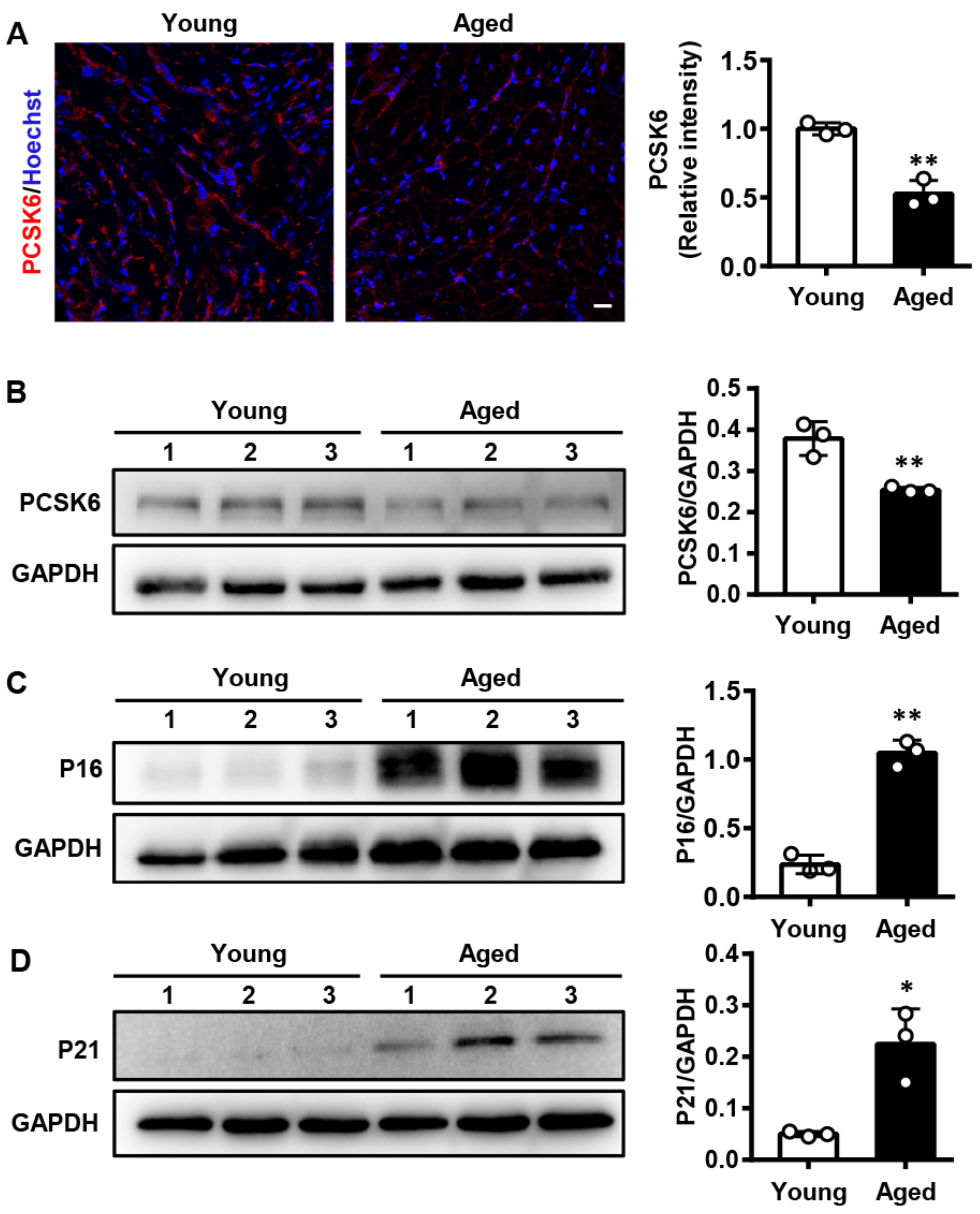
3.1. PCSK6 Protein Expression Is Reduced in the Heart of Aged Mice
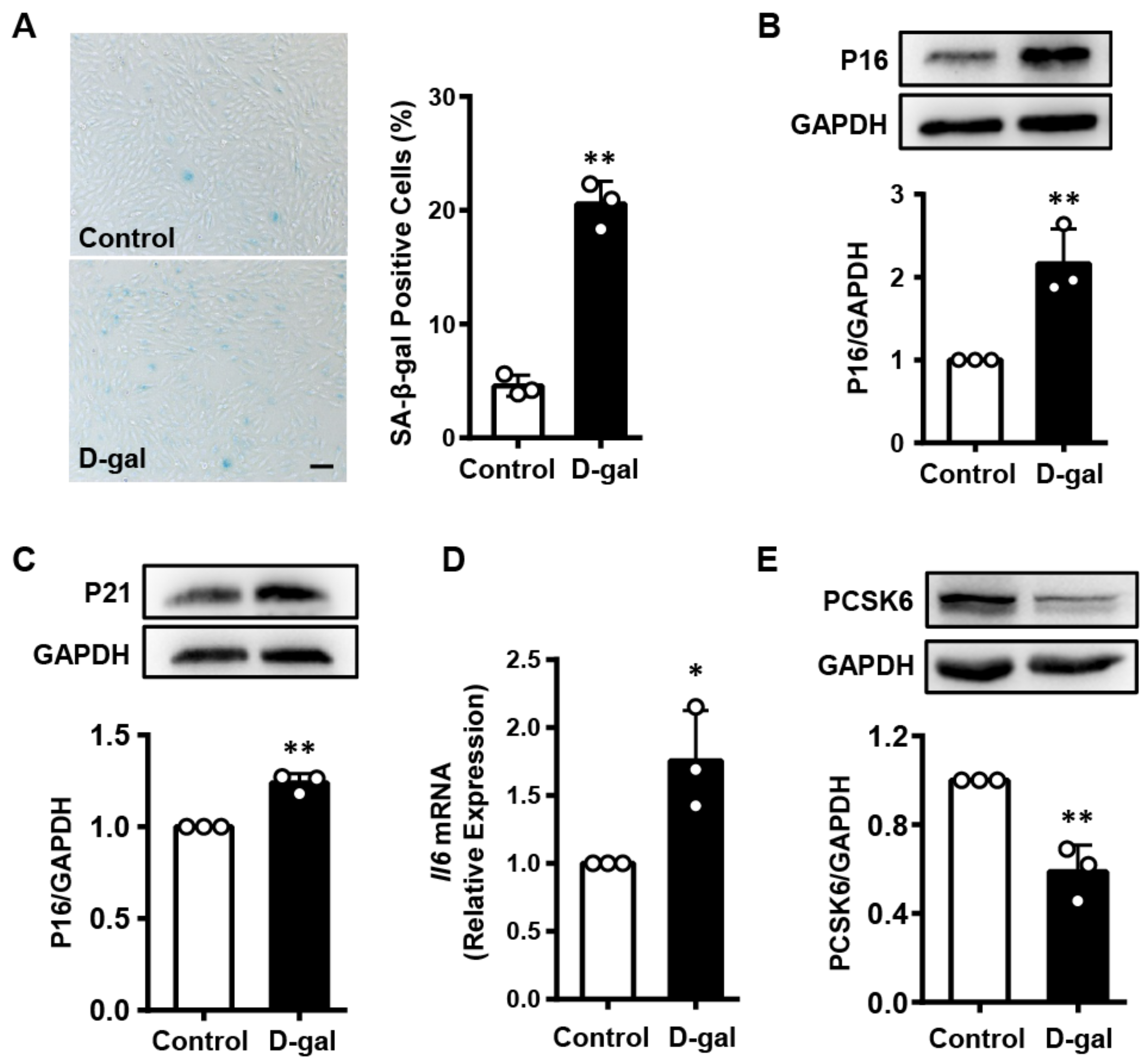
3.2. PCSK6 Expression Is Decreased in the Senescent Cardiomyocytes Induced by D-Gal
3.3. Pcsk6 Gene Knockdown Promotes Cardiomyocyte Senescence
3.4. Pcsk6 Gene Knockdown Impairs Cardiomyocyte Function
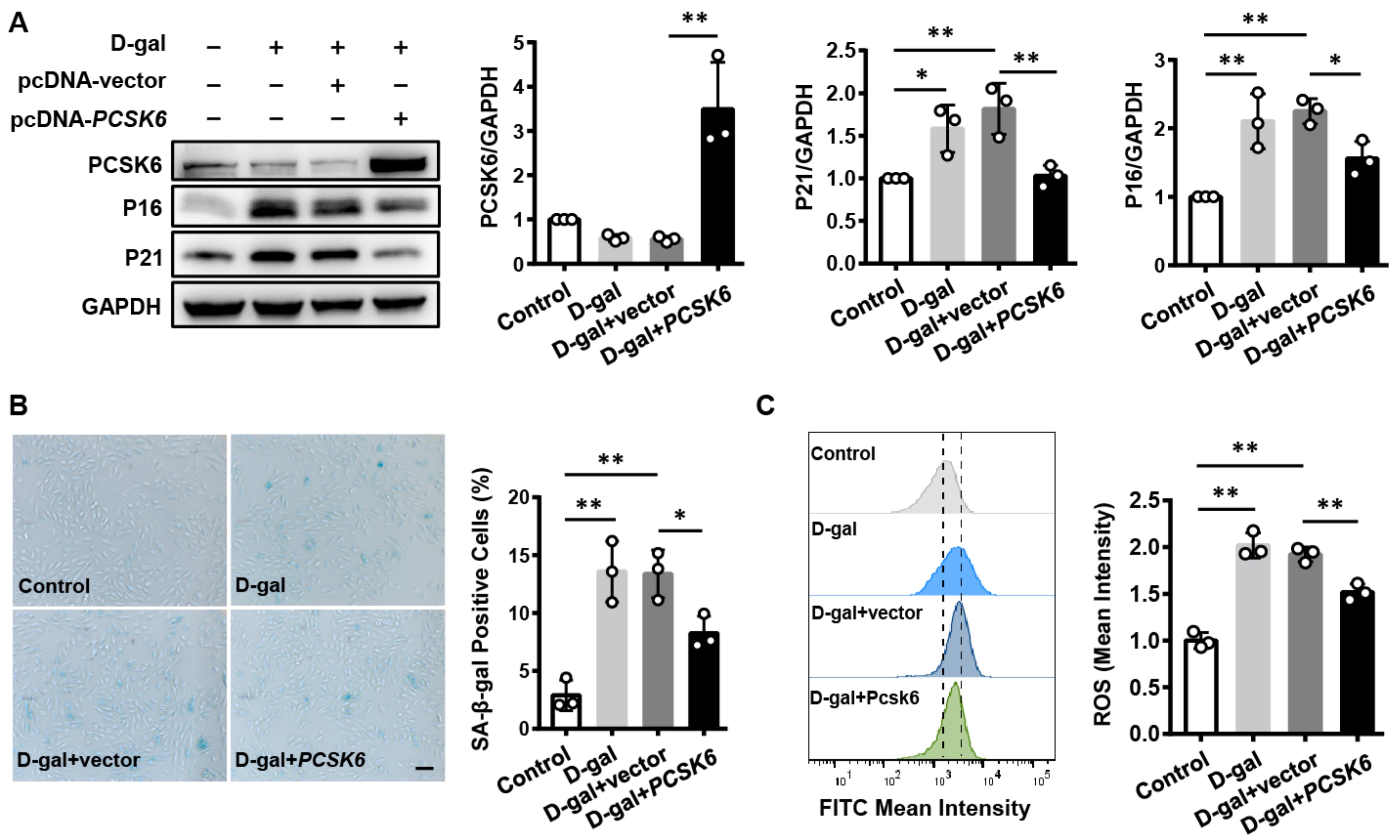
3.5. PCSK6 Overexpression Reverses Cardiomyocyte Senescence and Dysfunction Induced by D-gal
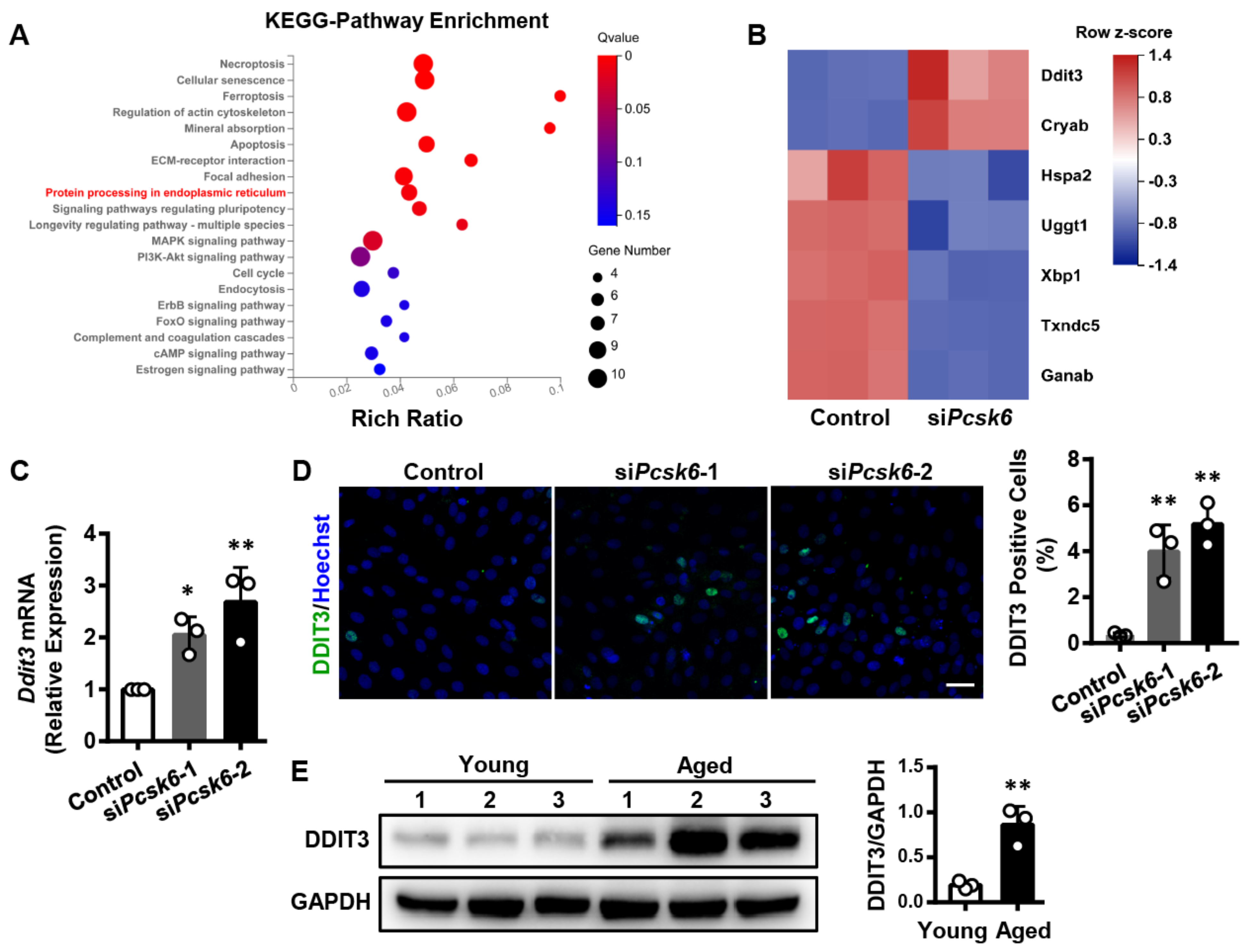
3.6. Identification of the Ddit3 Gene Related to Protein Processing in the Endoplasmic Reticulum (ER) upon Pcsk6-Knockdown
3.7. PCSK6 Regulates DDIT3 Expression in Tunicamycin (Tm)-Treated Cardiomyocytes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Johnson, S.C.; Szeto, H.; Rabinovitch, P.S. Cardiac aging: From molecular mechanisms to significance in human health and disease. Antioxid. Redox. Signal 2012, 16, 1492–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakou, E.S.; Parthenakis, F.I.; Kallergis, E.M.; Marketou, M.E.; Nakos, K.S.; Vardas, P.E. Healthy aging and myocardium: A complicated process with various effects in cardiac structure and physiology. Int. J. Cardiol. 2016, 209, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Steenman, M.; Lande, G. Cardiac aging and heart disease in humans. Biophys. Rev. 2017, 9, 131–137. [Google Scholar] [CrossRef]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Treguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Bernhard, D.; Laufer, G. The aging cardiomyocyte: A mini-review. Gerontology 2008, 54, 24–31. [Google Scholar] [CrossRef]
- Volpe, M. Natriuretic peptides and cardio-renal disease. Int. J. Cardiol. 2014, 176, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Wu, F.; Morser, J.; Wu, Q. Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proc. Natl. Acad. Sci. USA 2000, 97, 8525–8529. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Yan, W.; Pan, J.; Morser, J.; Wu, Q. Processing of pro-atrial natriuretic peptide by corin in cardiac myocytes. J. Biol. Chem. 2002, 277, 16900–16905. [Google Scholar] [CrossRef] [Green Version]
- Pollack, J.A.; Skvorak, J.P.; Nazian, S.J.; Landon, C.S.; Dietz, J.R. Alterations in atrial natriuretic peptide (ANP) secretion and renal effects in aging. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52, B196–B202. [Google Scholar] [CrossRef]
- Iemitsu, M.; Maeda, S.; Otsuki, T.; Sugawara, J.; Kuno, S.; Ajisaka, R.; Matsuda, M. Arterial stiffness, physical activity, and atrial natriuretic Peptide gene polymorphism in older subjects. Hypertens. Res. 2008, 31, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Häseli, S.; Deubel, S.; Jung, T.; Grune, T.; Ott, C. Cardiomyocyte Contractility and Autophagy in a Premature Senescence Model of Cardiac Aging. Oxid. Med. Cell. Longev. 2020, 2020, 8141307. [Google Scholar] [CrossRef]
- Chen, S.; Cao, P.; Dong, N.; Peng, J.; Zhang, C.; Wang, H.; Zhou, T.; Yang, J.; Zhang, Y.; Martelli, E.E.; et al. PCSK6-mediated corin activation is essential for normal blood pressure. Nat. Med. 2015, 21, 1048–1053. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, H.; Li, H.; Zhang, Y.; Wu, Q. Functional analysis of corin protein domains required for PCSK6-mediated activation. Int. J. Biochem. Cell. Biol. 2018, 94, 31–39. [Google Scholar] [CrossRef]
- Chen, S.; Sen, S.; Young, D.; Wang, W.; Moravec, C.S.; Wu, Q. Protease corin expression and activity in failing hearts. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1687–H1692. [Google Scholar] [CrossRef] [Green Version]
- Dong, N.; Chen, S.; Yang, J.; He, L.; Liu, P.; Zheng, D.; Li, L.; Zhou, Y.; Ruan, C.; Plow, E.; et al. Plasma soluble corin in patients with heart failure. Circ. Heart Fail. 2010, 3, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Constam, D.B.; Robertson, E.J. SPC4/PACE4 regulates a TGFbeta signaling network during axis formation. Genes Dev. 2000, 14, 1146–1155. [Google Scholar] [CrossRef]
- Seidah, N.G.; Sadr, M.S.; Chretien, M.; Mbikay, M. The multifaceted proprotein convertases: Their unique, redundant, complementary, and opposite functions. J. Biol. Chem. 2013, 288, 21473–21481. [Google Scholar] [CrossRef] [Green Version]
- Lyu, G.; Guan, Y.; Zhang, C.; Zong, L.; Sun, L.; Huang, X.; Huang, L.; Zhang, L.; Tian, X.L.; Zhou, Z.; et al. TGF-beta signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat. Commun. 2018, 9, 2560. [Google Scholar] [CrossRef]
- Rykaczewska, U.; Suur, B.E.; Rohl, S.; Razuvaev, A.; Lengquist, M.; Sabater-Lleal, M.; van der Laan, S.W.; Miller, C.L.; Wirka, R.C.; Kronqvist, M.; et al. PCSK6 Is a Key Protease in the Control of Smooth Muscle Cell Function in Vascular Remodeling. Circ. Res. 2020, 126, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, T.C.; Knobel, J.; Burkert-Rettenmaier, S.; Li, X.; Meyer, I.S.; Jungmann, A.; Sicklinger, F.; Backs, J.; Lasitschka, F.; Muller, O.J.; et al. Secretome Analysis of Cardiomyocytes Identifies PCSK6 as a Novel Player in Cardiac Remodeling after Myocardial Infarction. Circulation 2020, 141, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Sun, B.; Hong, Q.; Yan, J.; Mu, D.; Li, J.; Sheng, H.; Guo, H. PACE4 regulates apoptosis in human prostate cancer cells via endoplasmic reticulum stress and mitochondrial signaling pathways. Drug Des. Dev. Ther. 2015, 9, 5911–5923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, X.H.; Fan, D.X.; Zhang, Y.; Li, M.Q.; Wu, H.X.; Jin, L.P. PCSK6 regulated by LH inhibits the apoptosis of human granulosa cells via activin A and TGFβ2. J. Endocrinol. 2014, 222, 151–160. [Google Scholar] [CrossRef]
- Mujoomdar, M.L.; Hogan, L.M.; Parlow, A.F.; Nachtigal, M.W. Pcsk6 mutant mice exhibit progressive loss of ovarian function, altered gene expression, and formation of ovarian pathology. Reproduction 2011, 141, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, J.G., Jr.; Fray, J.; Leonard, J.L.; Pratt, R.E. Molecular mechanisms of reduced beta-adrenergic signaling in the aged heart as revealed by genomic profiling. Physiol. Genom. 2003, 15, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Zordoky, B.N.; El-Kadi, A.O. H9c2 cell line is a valuable in vitro model to study the drug metabolizing enzymes in the heart. J. Pharmacol. Toxicol. Methods 2007, 56, 317–322. [Google Scholar] [CrossRef]
- Tang, X.; Li, P.H.; Chen, H.Z. Cardiomyocyte Senescence and Cellular Communications within Myocardial Microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Vallentin, A.; Churchill, E.; Mochly-Rosen, D. deltaPKC participates in the endoplasmic reticulum stress-induced response in cultured cardiac myocytes and ischemic heart. J. Mol. Cell. Cardiol. 2007, 43, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Doll, S.; Dressen, M.; Geyer, P.E.; Itzhak, D.N.; Braun, C.; Doppler, S.A.; Meier, F.; Deutsch, M.A.; Lahm, H.; Lange, R.; et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 2017, 8, 1469. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Kerkela, R. Regulatory Mechanisms of Mitochondrial Function and Cardiac Aging. Int. J. Mol. Sci. 2020, 21, 1359. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.J.; Scheibye-Knudsen, M.; Longo, D.L.; de Cabo, R. Animal models of aging research: Implications for human aging and age-related diseases. Annu. Rev. Anim. Biosci. 2015, 3, 283–303. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.C.; Liu, J.H.; Wu, R.Y. Establishment of the mimetic aging effect in mice caused by D-galactose. Biogerontology 2003, 4, 15–18. [Google Scholar] [CrossRef]
- Wang, L.F.; Cao, Q.; Wen, K.; Xiao, Y.F.; Chen, T.T.; Guan, X.H.; Liu, Y.; Zuo, L.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Alleviates D-Galactose-Induced Myocardial Cell Senescence Through NAD(+)/Sirt1 Signaling Pathway. Front. Physiol. 2019, 10, 1125. [Google Scholar] [CrossRef] [Green Version]
- Maejima, Y.; Adachi, S.; Ito, H.; Hirao, K.; Isobe, M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell 2008, 7, 125–136. [Google Scholar] [CrossRef]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, F.; Preston, C.C.; Emelyanova, L.; Yousufuddin, M.; Viqar, M.; Dakwar, O.; Ross, G.R.; Faustino, R.S.; Holmuhamedov, E.L.; Jahangir, A. Effects of Aging on Cardiac Oxidative Stress and Transcriptional Changes in Pathways of Reactive Oxygen Species Generation and Clearance. J. Am. Heart Assoc. 2021, 10, e019948. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.J. Positive oxidative stress in aging and aging-related disease tolerance. Redox. Biol. 2014, 2, 165–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Shah, A.M. Age-associated pro-inflammatory remodeling and functional phenotype in the heart and large arteries. J. Mol. Cell Cardiol. 2015, 83, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosnowska, D.; Richardson, C.; Sonntag, W.E.; Csiszar, A.; Ungvari, Z.; Ridgway, I. A heart that beats for 500 years: Age-related changes in cardiac proteasome activity, oxidative protein damage and expression of heat shock proteins, inflammatory factors, and mitochondrial complexes in Arctica islandica, the longest-living noncolonial animal. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1448–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- No, M.H.; Choi, Y.; Cho, J.; Heo, J.W.; Cho, E.J.; Park, D.H.; Kang, J.H.; Kim, C.J.; Seo, D.Y.; Han, J.; et al. Aging Promotes Mitochondria-Mediated Apoptosis in Rat Hearts. Life 2020, 10, 178. [Google Scholar] [CrossRef]
- Song, X.; Bao, M.; Li, D.; Li, Y.M. Advanced glycation in D-galactose induced mouse aging model. Mech. Ageing Dev. 1999, 108, 239–251. [Google Scholar] [CrossRef]
- Simm, A.; Müller, B.; Nass, N.; Hofmann, B.; Bushnaq, H.; Silber, R.E.; Bartling, B. Protein glycation—Between tissue aging and protection. Exp. Gerontol. 2015, 68, 71–75. [Google Scholar] [CrossRef]
- Zieman, S.; Kass, D. Advanced glycation end product cross-linking: Pathophysiologic role and therapeutic target in cardiovascular disease. Congest. Heart Fail. 2004, 10, 144–149. [Google Scholar] [CrossRef]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Lu, Q.; Hu, Z.; Yu, Y.; Chen, Q.; Wang, Q.K. A non-canonical pathway regulates ER stress signaling and blocks ER stress-induced apoptosis and heart failure. Nat. Commun. 2017, 8, 133. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, M.; Meijering, R.A.M.; Qi, X.Y.; Zhang, D.; Liu, T.; Hoogstra-Berends, F.; Sibon, O.C.M.; Henning, R.H.; Nattel, S.; Brundel, B. Endoplasmic Reticulum Stress Is Associated with Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Heart Assoc. 2017, 6, e006458. [Google Scholar] [CrossRef] [Green Version]
- Dally, S.; Monceau, V.; Corvazier, E.; Bredoux, R.; Raies, A.; Bobe, R.; del Monte, F.; Enouf, J. Compartmentalized expression of three novel sarco/endoplasmic reticulum Ca2+ATPase 3 isoforms including the switch to ER stress, SERCA3f, in non-failing and failing human heart. Cell Calcium 2009, 45, 144–154. [Google Scholar] [CrossRef]
- Chen, Q.; Samidurai, A.; Thompson, J.; Hu, Y.; Das, A.; Willard, B.; Lesnefsky, E.J. Endoplasmic reticulum stress-mediated mitochondrial dysfunction in aged hearts. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165899. [Google Scholar] [CrossRef]
- Nerlov, C. The C/EBP family of transcription factors: A paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007, 17, 318–324. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.Y.; Okada, K.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Sawada, T.; Okuda, K.; Asano, Y.; Sanada, S.; et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361–369. [Google Scholar] [CrossRef]
- Hamada, H.; Suzuki, M.; Yuasa, S.; Mimura, N.; Shinozuka, N.; Takada, Y.; Suzuki, M.; Nishino, T.; Nakaya, H.; Koseki, H.; et al. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol. Cell Biol. 2004, 24, 8007–8017. [Google Scholar] [CrossRef] [Green Version]
- Maytin, E.V.; Ubeda, M.; Lin, J.C.; Habener, J.F. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp. Cell Res. 2001, 267, 193–204. [Google Scholar] [CrossRef]
- Isodono, K.; Takahashi, T.; Imoto, H.; Nakanishi, N.; Ogata, T.; Asada, S.; Adachi, A.; Ueyama, T.; Oh, H.; Matsubara, H. PARM-1 is an endoplasmic reticulum molecule involved in endoplasmic reticulum stress-induced apoptosis in rat cardiac myocytes. PLoS ONE 2010, 5, e9746. [Google Scholar] [CrossRef]
- Ochoa, C.D.; Wu, R.F.; Terada, L.S. ROS signaling and ER stress in cardiovascular disease. Mol. Asp. Med. 2018, 63, 18–29. [Google Scholar] [CrossRef]
- Abhari, B.A.; McCarthy, N.; Le Berre, M.; Kilcoyne, M.; Joshi, L.; Agostinis, P.; Fulda, S. Smac mimetic suppresses tunicamycin-induced apoptosis via resolution of ER stress. Cell Death Dis. 2019, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.; Hassanzadeh, S.; Singh, K.; Menazza, S.; Nguyen, T.T.; Stevens, M.V.; Nguyen, A.; San, H.; Anderson, S.A.; Lin, Y.; et al. Parkin regulation of CHOP modulates susceptibility to cardiac endoplasmic reticulum stress. Sci. Rep. 2017, 7, 2093. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Abdullah, C.S.; Aishwarya, R.; Orr, A.W.; Traylor, J.; Miriyala, S.; Panchatcharam, M.; Pattillo, C.B.; Bhuiyan, M.S. Sigmar1 regulates endoplasmic reticulum stress-induced C/EBP-homologous protein expression in cardiomyocytes. Biosci. Rep. 2017, 37, BSR20170898. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, W.; Chen, L.; Liu, H.; Long, C.; Liu, J.; Ding, S.; Wu, Q.; Chen, S. Pcsk6 Deficiency Promotes Cardiomyocyte Senescence by Modulating Ddit3-Mediated ER Stress. Genes 2022, 13, 711. https://doi.org/10.3390/genes13040711
Zhan W, Chen L, Liu H, Long C, Liu J, Ding S, Wu Q, Chen S. Pcsk6 Deficiency Promotes Cardiomyocyte Senescence by Modulating Ddit3-Mediated ER Stress. Genes. 2022; 13(4):711. https://doi.org/10.3390/genes13040711
Chicago/Turabian StyleZhan, Wenxing, Liping Chen, Hongfei Liu, Changkun Long, Jiankun Liu, Shuangjin Ding, Qingyu Wu, and Shenghan Chen. 2022. "Pcsk6 Deficiency Promotes Cardiomyocyte Senescence by Modulating Ddit3-Mediated ER Stress" Genes 13, no. 4: 711. https://doi.org/10.3390/genes13040711
APA StyleZhan, W., Chen, L., Liu, H., Long, C., Liu, J., Ding, S., Wu, Q., & Chen, S. (2022). Pcsk6 Deficiency Promotes Cardiomyocyte Senescence by Modulating Ddit3-Mediated ER Stress. Genes, 13(4), 711. https://doi.org/10.3390/genes13040711