
MICAL1 Monooxygenase in Autosomal Dominant Lateral Temporal Epilepsy: Role in Cytoskeletal Regulation and Relation to Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Clinical Characteristics and Genetic Transmission of ADLTE
2. LGI1, Reelin and MICAL1 Genes in the Etiology of ADLTE
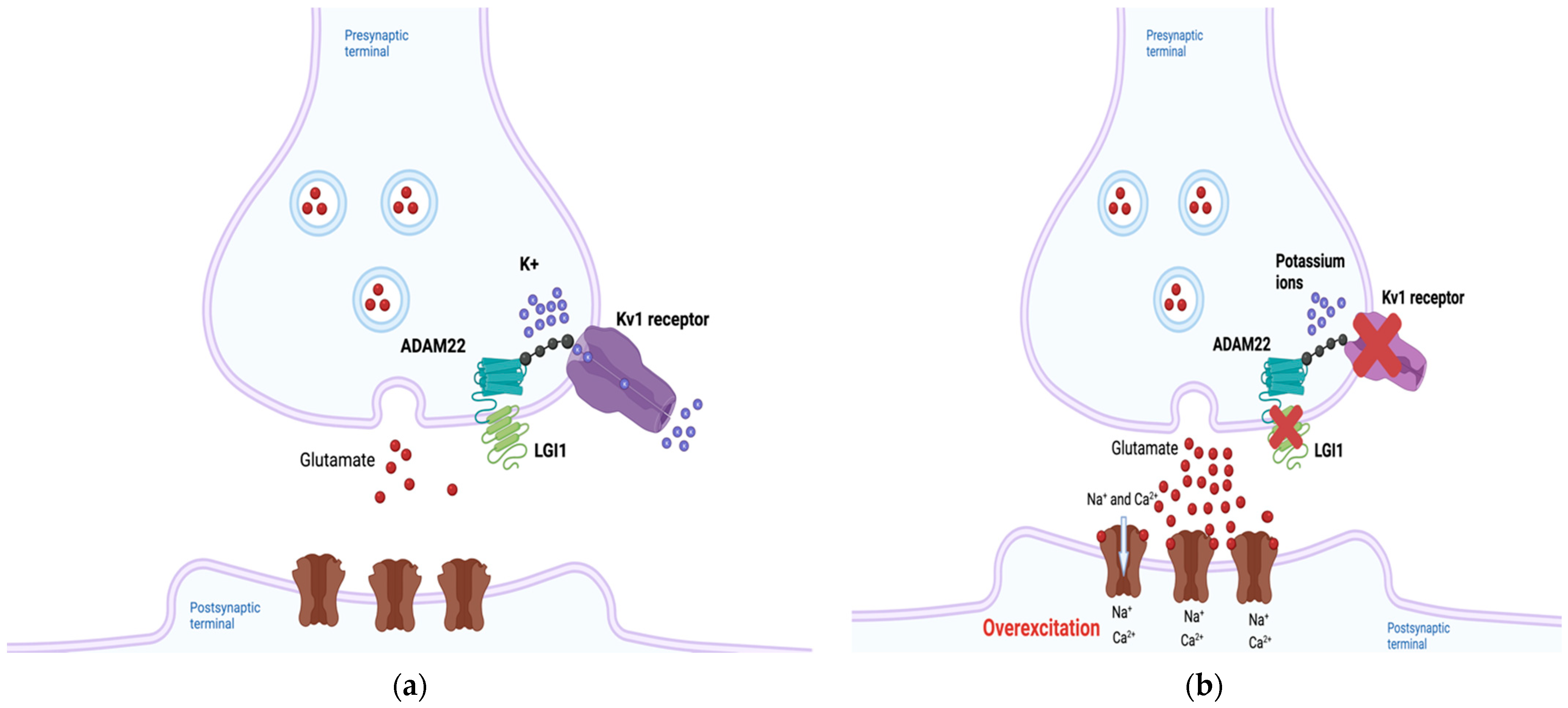
2.1. LGI1
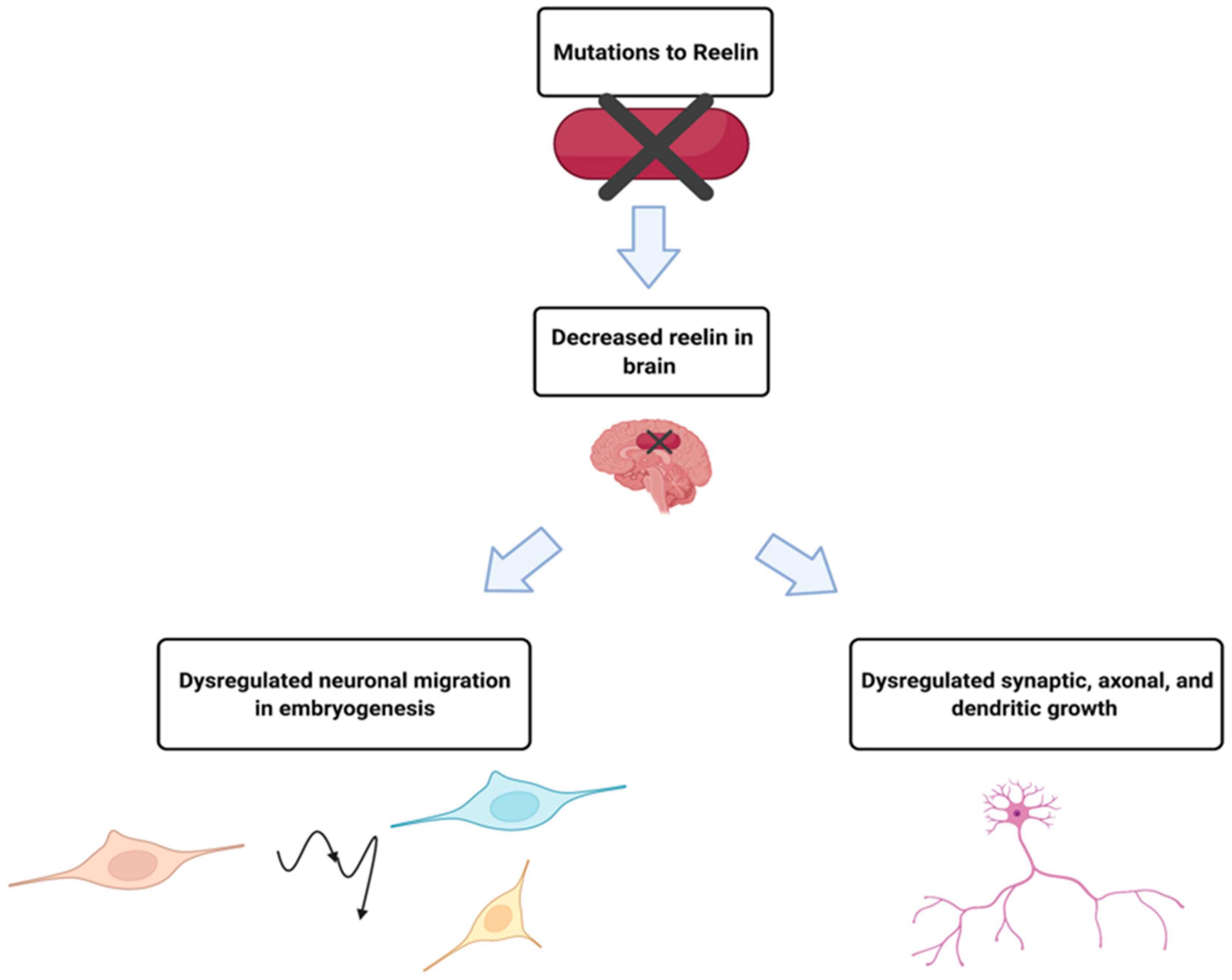
2.2. RELN
2.3. MICAL1
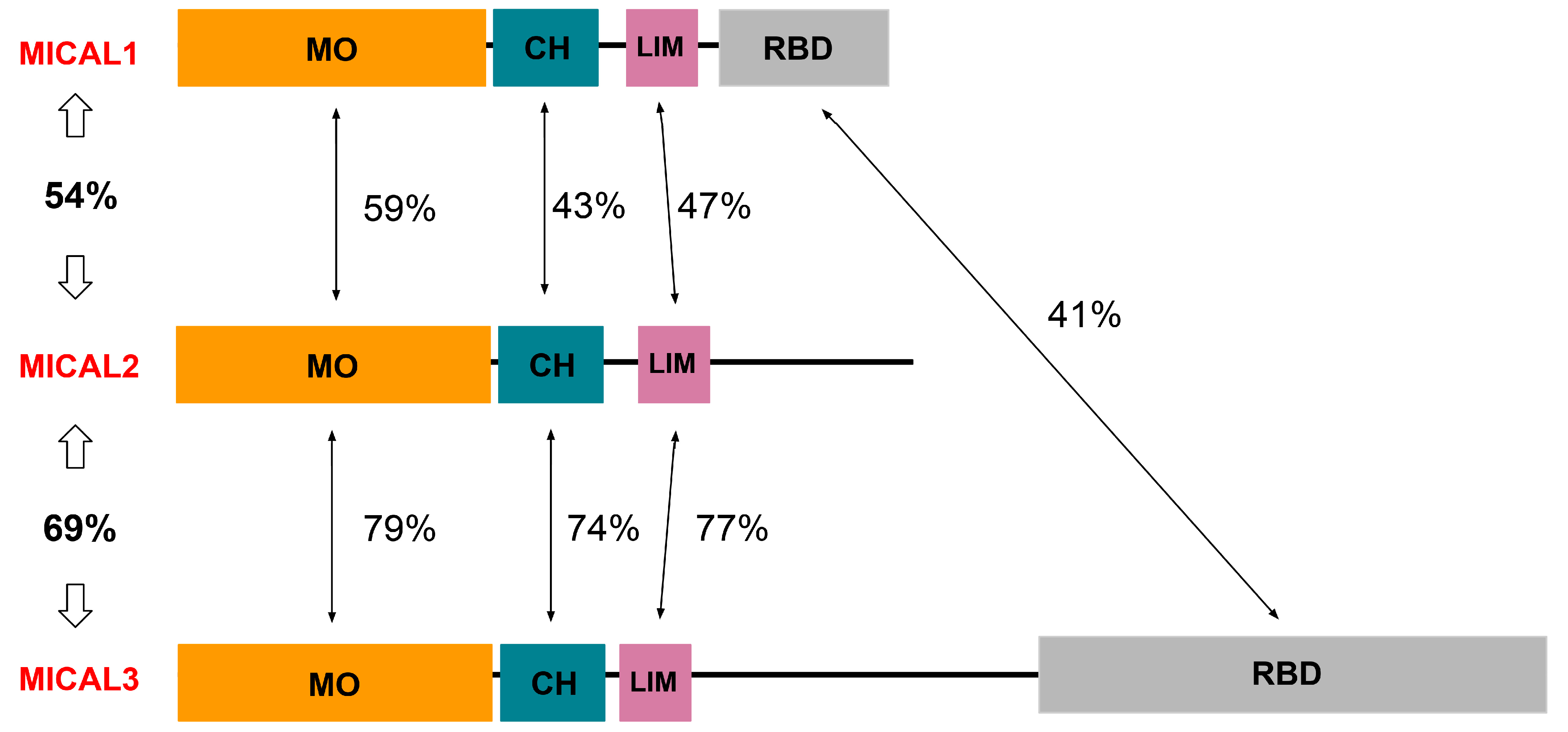
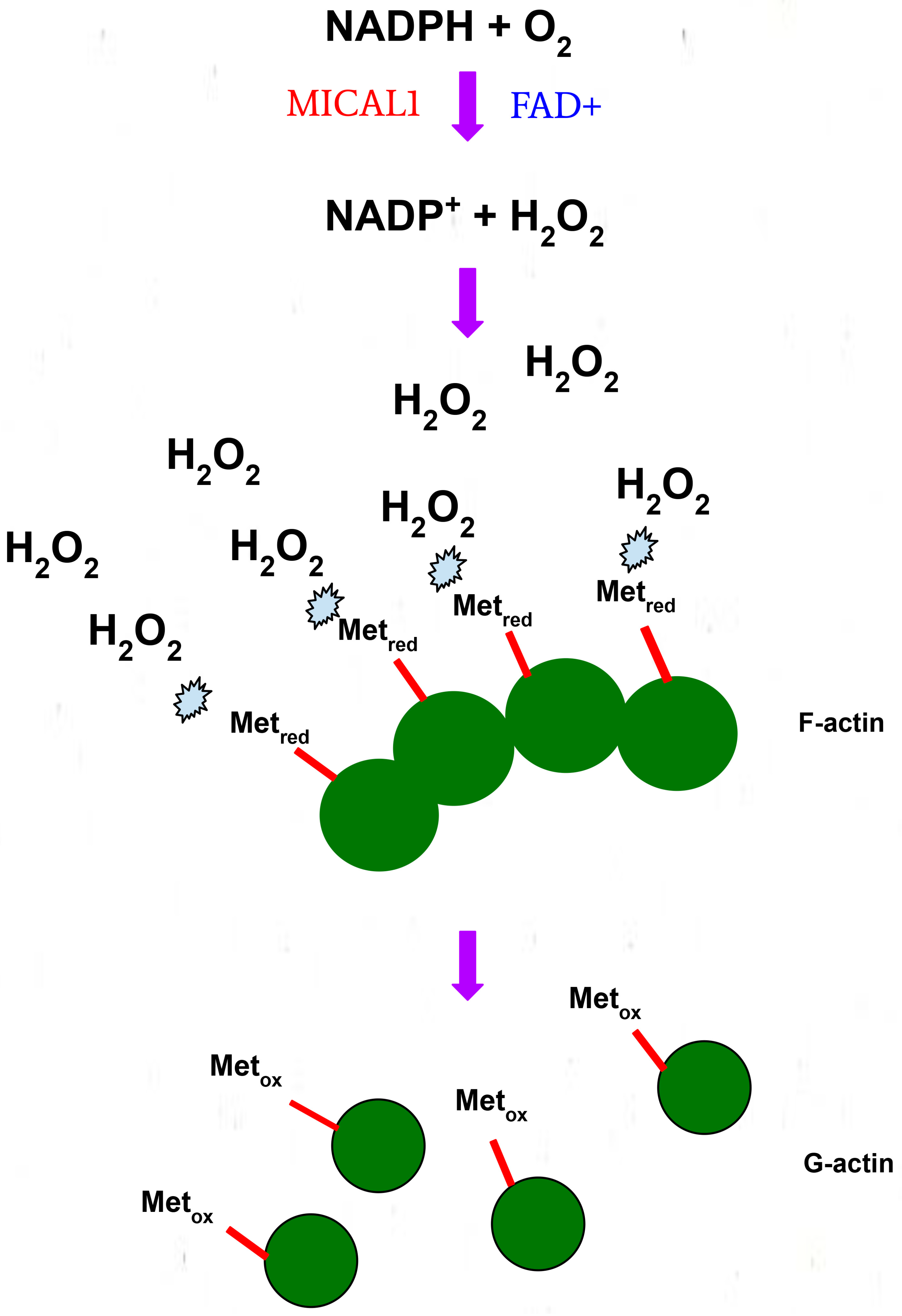
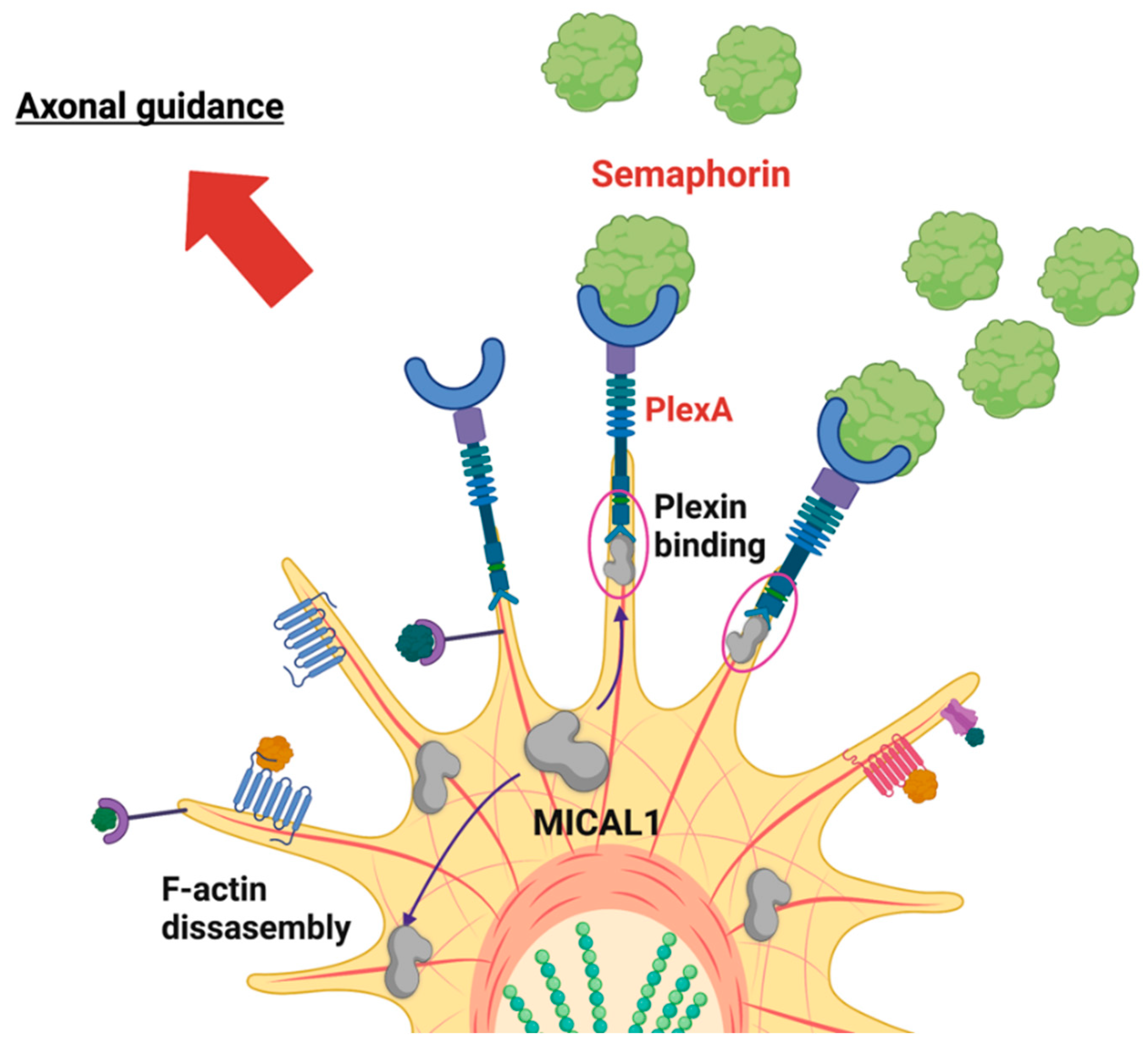
3. Structure and Function of MICAL1
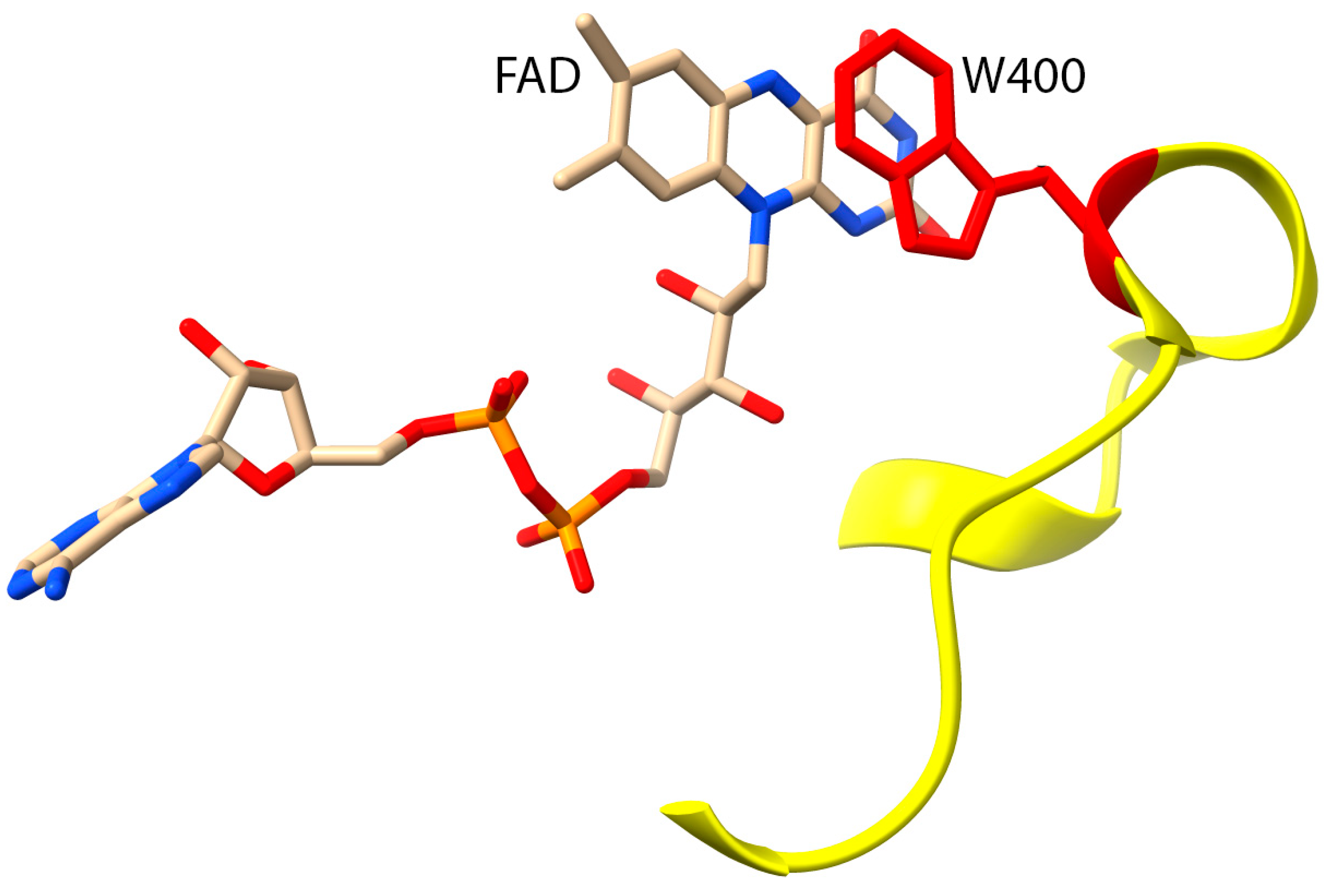
4. Biochemical Roles of G150 and A1065
5. MICAL1 in Central Nervous System Biology and ADLTE Etiology
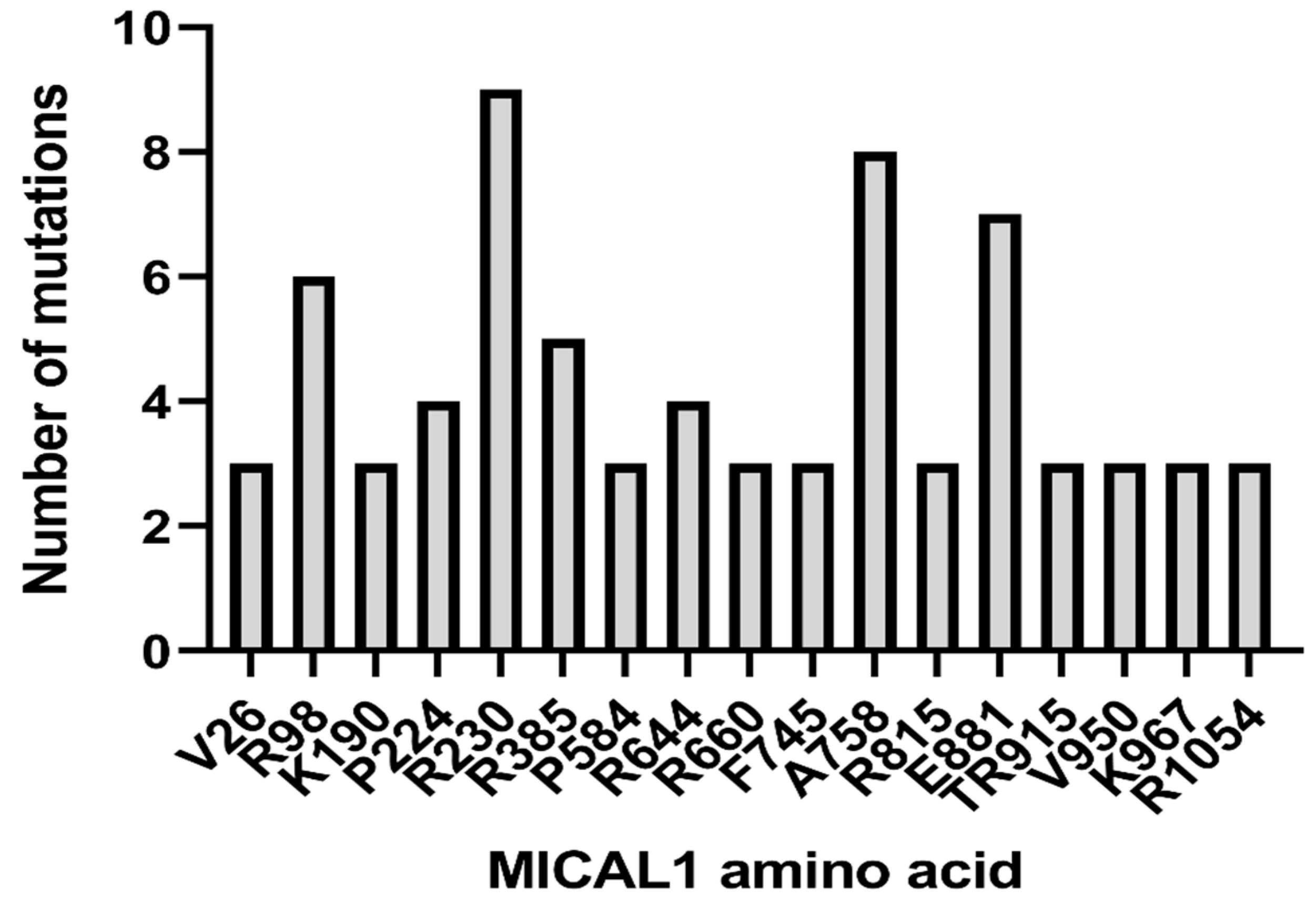
6. ADLTE-Associated MICAL1 Mutations in Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Michelucci, R.; Pulitano, P.; Di Bonaventura, C.; Binelli, S.; Luisi, C.; Pasini, E.; Striano, S.; Striano, P.; Coppola, G.; La Neve, A.; et al. The Clinical Phenotype of Autosomal Dominant Lateral Temporal Lobe Epilepsy Related to Reelin Mutations. Epilepsy Behav. 2017, 68, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Stafstrom, C.E.; Carmant, L. Seizures and Epilepsy: An Overview for Neuroscientists. Cold Spring Harb. Perspect. Med. 2015, 5, a022426. [Google Scholar] [CrossRef] [PubMed]
- Dazzo, E.; Rehberg, K.; Michelucci, R.; Passarelli, D.; Boniver, C.; Vianello Dri, V.; Striano, P.; Striano, S.; Pasterkamp, R.J.; Nobile, C. Mutations in MICAL-1 Cause Autosomal-Dominant Lateral Temporal Epilepsy: MICAL-1 Mutations in ADLTE. Ann. Neurol. 2018, 83, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Dazzo, E.; Fanciulli, M.; Serioli, E.; Minervini, G.; Pulitano, P.; Binelli, S.; Di Bonaventura, C.; Luisi, C.; Pasini, E.; Striano, S.; et al. Heterozygous Reelin Mutations Cause Autosomal-Dominant Lateral Temporal Epilepsy. Am. J. Hum. Genet. 2015, 96, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Dazzo, E.; Santulli, L.; Posar, A.; Fattouch, J.; Conti, S.; Lodén-van Straaten, M.; Mijalkovic, J.; De Bortoli, M.; Rosa, M.; Millino, C.; et al. Autosomal Dominant Lateral Temporal Epilepsy (ADLTE): Novel Structural and Single-Nucleotide LGI1 Mutations in Families with Predominant Visual Auras. Epilepsy Res. 2015, 110, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, R.; Pasini, E.; Malacrida, S.; Striano, P.; Bonaventura, C.D.; Pulitano, P.; Bisulli, F.; Egeo, G.; Santulli, L.; Sofia, V.; et al. Low Penetrance of Autosomal Dominant Lateral Temporal Epilepsy in Italian Families without LGI1 Mutations. Epilepsia 2013, 54, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, R.; Nobile, C. Autosomal Dominant Epilepsy with Auditory Features. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Ottman, R.; Winawer, M.R.; Kalachikov, S.; Barker-Cummings, C.; Gilliam, T.C.; Pedley, T.A.; Hauser, W.A. LGI1 Mutations in Autosomal Dominant Partial Epilepsy with Auditory Features. Neurology 2004, 62, 1120–1126. [Google Scholar] [CrossRef] [Green Version]
- Ottman, R.; Risch, N.; Hauser, W.A.; Pedley, T.A.; Lee, J.H.; Barker-Cummings, C.; Lustenberger, A.; Nagle, K.J.; Lee, K.S.; Scheuer, M.L.; et al. Localization of a Gene for Partial Epilepsy to Chromosome 10q. Nat. Genet. 1995, 10, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Seagar, M.; Russier, M.; Caillard, O.; Maulet, Y.; Fronzaroli-Molinieres, L.; De San Feliciano, M.; Boumedine-Guignon, N.; Rodriguez, L.; Zbili, M.; Usseglio, F.; et al. LGI1 Tunes Intrinsic Excitability by Regulating the Density of Axonal Kv1 Channels. Proc. Natl. Acad. Sci. USA 2017, 114, 7719–7724. [Google Scholar] [CrossRef] [Green Version]
- Baudin, P.; Cousyn, L.; Navarro, V. The LGI1 Protein: Molecular Structure, Physiological Functions and Disruption-Related Seizures. Cell. Mol. Life Sci. 2022, 79, 16. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Hirano, Y.; Miyazaki, Y.; Yokoi, N.; Fukata, M. Trans-Synaptic LGI1–ADAM22–MAGUK in AMPA and NMDA Receptor Regulation. Neuropharmacology 2021, 194, 108628. [Google Scholar] [CrossRef] [PubMed]
- Schulte, U.; Thumfart, J.-O.; Klöcker, N.; Sailer, C.A.; Bildl, W.; Biniossek, M.; Dehn, D.; Deller, T.; Eble, S.; Abbass, K.; et al. The Epilepsy-Linked Lgi1 Protein Assembles into Presynaptic Kv1 Channels and Inhibits Inactivation by Kvβ1. Neuron 2006, 49, 697–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, Y.; Chen, X.; Chiken, S.; Hirano, Y.; Yamagata, A.; Inahashi, H.; Sanbo, M.; Sano, H.; Goto, T.; Hirabayashi, M.; et al. LGI1–ADAM22–MAGUK Configures Transsynaptic Nanoalignment for Synaptic Transmission and Epilepsy Prevention. Proc. Natl. Acad. Sci. USA. 2021, 118, e2022580118. [Google Scholar] [CrossRef]
- Yamagata, A.; Miyazaki, Y.; Yokoi, N.; Shigematsu, H.; Sato, Y.; Goto-Ito, S.; Maeda, A.; Goto, T.; Sanbo, M.; Hirabayashi, M.; et al. Structural Basis of Epilepsy-Related Ligand–Receptor Complex LGI1–ADAM22. Nat. Commun. 2018, 9, 1546. [Google Scholar] [CrossRef]
- Yamagata, A.; Fukai, S. Insights into the Mechanisms of Epilepsy from Structural Biology of LGI1–ADAM22. Cell. Mol. Life Sci. 2020, 77, 267–274. [Google Scholar] [CrossRef]
- Zhou, Y.-D.; Lee, S.; Jin, Z.; Wright, M.; Smith, S.E.P.; Anderson, M.P. Arrested Maturation of Excitatory Synapses in Autosomal Dominant Lateral Temporal Lobe Epilepsy. Nat. Med. 2009, 15, 1208–1214. [Google Scholar] [CrossRef]
- Anderson, M.P. Arrested Glutamatergic Synapse Development in Human Partial Epilepsy. Epilepsy Curr. 2010, 10, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Senechal, K.R.; Thaller, C.; Noebels, J.L. ADPEAF Mutations Reduce Levels of Secreted LGI1, a Putative Tumor Suppressor Protein Linked to Epilepsy. Hum. Mol. Genet. 2005, 14, 1613–1620. [Google Scholar] [CrossRef] [Green Version]
- Dazzo, E.; Leonardi, E.; Belluzzi, E.; Malacrida, S.; Vitiello, L.; Greggio, E.; Tosatto, S.C.E.; Nobile, C. Secretion-Positive LGI1 Mutations Linked to Lateral Temporal Epilepsy Impair Binding to ADAM22 and ADAM23 Receptors. PLoS Genet. 2016, 12, e1006376. [Google Scholar] [CrossRef]
- Rosanoff, M.J.; Ottman, R. Penetrance of LGI1 Mutations in Autosomal Dominant Partial Epilepsy with Auditory Features. Neurology 2008, 71, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Wu, D.; Zang, Y.; Wang, Y.; Zhou, Y.; Qiao, F.; Teng, X.; Chen, J.; Li, Q.; Sun, J.; et al. A Novel LGI1 Mutation Causing Autosomal Dominant Lateral Temporal Lobe Epilepsy Confirmed by a Precise Knock-in Mouse Model. CNS Neurosci. Ther. 2022, 28, 237–246. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G. Reelin in the Years: Controlling Neuronal Migration and Maturation in the Mammalian Brain. Adv. Neurosci. 2014, 2014, 597395. [Google Scholar] [CrossRef]
- Dazzo, E.; Nobile, C. Epilepsy-Causing Reelin Mutations Result in Impaired Secretion and Intracellular Degradation of Mutant Proteins. Hum. Mol. Genet. 2021, 31, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Fanciulli, M.; Pasini, E.; Malacrida, S.; Striano, P.; Striano, S.; Michelucci, R.; Ottman, R.; Nobile, C. Copy Number Variations and Susceptibility to Lateral Temporal Epilepsy: A Study of 21 Pedigrees. Epilepsia 2014, 55, 1651–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisulli, F.; Naldi, I.; Baldassari, S.; Magini, P.; Licchetta, L.; Castegnaro, G.; Fabbri, M.; Stipa, C.; Ferrari, S.; Seri, M.; et al. Autosomal Dominant Partial Epilepsy with Auditory Features: A New Locus on Chromosome 19q13.11-q13.31. Epilepsia 2014, 55, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Terman, J.R.; Mao, T.; Pasterkamp, R.J.; Yu, H.-H.; Kolodkin, A.L. MICALs, a Family of Conserved Flavoprotein Oxidoreductases, Function in Plexin-Mediated Axonal Repulsion. Cell 2002, 109, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, S.S.P.; Caplan, S. MICAL-Family Proteins: Complex Regulators of the Actin Cytoskeleton. Antioxid. Redox Signal. 2014, 20, 2059–2073. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, S.S.P.; Rohn, J.L.; Naslavsky, N.; Caplan, S. Differential Regulation of Actin Microfilaments by Human MICAL Proteins. J. Cell Sci. 2012, 125, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Pasterkamp, R.J.; Dai, H.; Terman, J.R.; Wahlin, K.J.; Kim, B.; Bregman, B.S.; Popovich, P.G.; Kolodkin, A.L. MICAL Flavoprotein Monooxygenases: Expression during Neural Development and Following Spinal Cord Injuries in the Rat. Mol. Cell. Neurosci. 2006, 31, 52–69. [Google Scholar] [CrossRef]
- Wu, H.; Yesilyurt, H.G.; Yoon, J.; Terman, J.R. The MICALs Are a Family of F-Actin Dismantling Oxidoreductases Conserved from Drosophila to Humans. Sci. Rep. 2018, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Brondani, P.B.; Dudek, H.M.; Martinoli, C.; Mattevi, A.; Fraaije, M.W. Finding the Switch: Turning a Baeyer–Villiger Monooxygenase into a NADPH Oxidase. J. Am. Chem. Soc. 2014, 136, 16966–16969. [Google Scholar] [CrossRef] [PubMed]
- Palfey, B.A.; McDonald, C.A. Control of Catalysis in Flavin-Dependent Monooxygenases. Arch. Biochem. Biophys. 2010, 493, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, M.A. Structure-Function Studies of MICAL, the Unusual Multidomain Flavoenzyme Involved in Actin Cytoskeleton Dynamics. Arch. Biochem. Biophys. 2017, 632, 118–141. [Google Scholar] [CrossRef]
- Zucchini, D.; Caprini, G.; Pasterkamp, R.J.; Tedeschi, G.; Vanoni, M.A. Kinetic and Spectroscopic Characterization of the Putative Monooxygenase Domain of Human MICAL-1. Arch. Biochem. Biophys. 2011, 515, 1–13. [Google Scholar] [CrossRef]
- Vitali, T.; Maffioli, E.; Tedeschi, G.; Vanoni, M.A. Properties and Catalytic Activities of MICAL1, the Flavoenzyme Involved in Cytoskeleton Dynamics, and Modulation by Its CH, LIM and C-terminal Domains. Arch. Biochem. Biophys. 2016, 593, 24–37. [Google Scholar] [CrossRef]
- Hung, R.-J.; Pak, C.W.; Terman, J.R. Direct Redox Regulation of F-Actin Assembly and Disassembly by Mical. Science 2011, 334, 1710–1713. [Google Scholar] [CrossRef] [Green Version]
- Huijbers, M.M.E.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavin Dependent Monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef]
- Siebold, C.; Berrow, N.; Walter, T.S.; Harlos, K.; Owens, R.J.; Stuart, D.I.; Terman, J.R.; Kolodkin, A.L.; Pasterkamp, R.J.; Jones, E.Y. High-Resolution Structure of the Catalytic Region of MICAL (Molecule Interacting with CasL), a Multidomain Flavoenzyme-Signaling Molecule. Proc. Natl. Acad. Sci. USA. 2005, 102, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Alqassim, S.S.; Urquiza, M.; Borgnia, E.; Nagib, M.; Amzel, L.M.; Bianchet, M.A. Modulation of MICAL Monooxygenase Activity by Its Calponin Homology Domain: Structural and Mechanistic Insights. Sci. Rep. 2016, 6, 22176. [Google Scholar] [CrossRef] [Green Version]
- Nadella, M.; Bianchet, M.A.; Gabelli, S.B.; Barrila, J.; Amzel, L.M. Structure and Activity of the Axon Guidance Protein MICAL. Proc. Natl. Acad. Sci. USA 2005, 102, 16830–16835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Zhang, J.; Dai, H.; Sun, H.; Wang, D.; Wu, J.; Shi, Y. Investigation of the Four Cooperative Unfolding Units Existing in the MICAL-1 CH Domain. Biophys. Chem. 2007, 129, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Kadrmas, J.L.; Beckerle, M.C. The LIM Domain: From the Cytoskeleton to the Nucleus. Nat. Rev. Mol. Cell Biol. 2004, 5, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.F.; Shim, S.-O.; Strittmatter, S.M. Release of MICAL Autoinhibition by Semaphorin-Plexin Signaling Promotes Interaction with Collapsin Response Mediator Protein. J. Neurosci. 2008, 28, 2287–2297. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.; Roh, Y.J.; Kim, H.-U.; Shin, D.; Kim, S.; Son, J.; Lee, A.; Kim, M.; Park, J.; et al. Structural and Kinetic Insights into Flavin-Containing Monooxygenase and Calponin-Homology Domains in Human MICAL3. IUCrJ 2020, 7, 90–99. [Google Scholar] [CrossRef]
- Tessier-Lavigne, M.; Goodman, C.S. The Molecular Biology of Axon Guidance. Science 1996, 274, 1123–1133. [Google Scholar] [CrossRef] [Green Version]
- Alto, L.T.; Terman, J.R. Semaphorins and Their Signaling Mechanisms. In Semaphorin Signaling; Terman, J.R., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1493, pp. 1–25. ISBN 978-1-4939-6446-8. [Google Scholar]
- Janssen, B.J.C.; Robinson, R.A.; Pérez-Brangulí, F.; Bell, C.H.; Mitchell, K.J.; Siebold, C.; Jones, E.Y. Structural Basis of Semaphorin–Plexin Signalling. Nature 2010, 467, 1118–1122. [Google Scholar] [CrossRef]
- Kahn, O.I.; Baas, P.W. Microtubules and Growth Cones: Motors Drive the Turn. Trends Neurosci. 2016, 39, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Morinaka, A.; Yamada, M.; Itofusa, R.; Funato, Y.; Yoshimura, Y.; Nakamura, F.; Yoshimura, T.; Kaibuchi, K.; Goshima, Y.; Hoshino, M.; et al. Thioredoxin Mediates Oxidation-Dependent Phosphorylation of CRMP2 and Growth Cone Collapse. Sci. Signal. 2011, 4, ra26. [Google Scholar] [CrossRef]
- Schmidt, E.F.; Strittmatter, S.M. The CRMP Family of Proteins and Their Role in Sema3A Signaling. In Semaphorins: Receptor and Intracellular Signaling Mechanisms; Pasterkamp, R.J., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2007; Volume 600, pp. 1–11. ISBN 978-0-387-70955-0. [Google Scholar]
- Zhou, F.; Cohan, C.S. Growth Cone Collapse through Coincident Loss of Actin Bundles and Leading Edge Actin without Actin Depolymerization. J. Cell Biol. 2001, 153, 1071–1084. [Google Scholar] [CrossRef] [Green Version]
- Turner, L.J.; Hall, A. Plexin-Induced Collapse Assay in COS Cells. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2006; Volume 406, pp. 665–676. ISBN 978-0-12-182811-0. [Google Scholar]
- Bamburg, J.R. Proteins of the ADF/Cofilin Family: Essential Regulators of Actin Dynamics. Annu. Rev. Cell Dev. Biol. 1999, 15, 185–230. [Google Scholar] [CrossRef] [PubMed]
- McGarry, D.J.; Armstrong, G.; Castino, G.; Mason, S.; Clark, W.; Shaw, R.; McGarry, L.; Blyth, K.; Olson, M.F. MICAL1 Regulates Actin Cytoskeleton Organization, Directional Cell Migration and the Growth of Human Breast Cancer Cells as Orthotopic Xenograft Tumours. Cancer Lett. 2021, 519, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, Y.; Gu, L.; Duan, B.; Cui, J.; Zhang, Y.; Chen, Y.; Sun, S.; Dong, J.; Du, J. MICAL1 Controls Cell Invasive Phenotype via Regulating Oxidative Stress in Breast Cancer Cells. BMC Cancer 2016, 16, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Min, P.; Liu, L.; Zhang, L.; Zhang, Y.; Wang, Y.; Zhao, X.; Ma, Y.; Xie, H.; Zhu, C.; et al. NEDD9 Facilitates Hypoxia-Induced Gastric Cancer Cell Migration via MICAL1 Related Rac1 Activation. Front. Pharmacol. 2019, 10, 291. [Google Scholar] [CrossRef] [Green Version]
- Loria, R.; Bon, G.; Perotti, V.; Gallo, E.; Bersani, I.; Baldassari, P.; Porru, M.; Leonetti, C.; Di Carlo, S.; Visca, P.; et al. Sema6A and Mical1 Control Cell Growth and Survival of BRAFV600E Human Melanoma Cells. Oncotarget 2015, 6, 2779–2793. [Google Scholar] [CrossRef] [Green Version]
- Chiang, A.C.; Massagué, J. Molecular Basis of Metastasis. N. Engl. J. Med. 2008, 359, 2814–2823. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.F.; Sahai, E. The Actin Cytoskeleton in Cancer Cell Motility. Clin. Exp. Metastasis 2009, 26, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.-Y.; Storz, P. Reactive Oxygen Species in Cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Kundu, N.; Zhang, S.; Fulton, A.M. Sublethal Oxidative Stress Inhibits Tumor Cell Adhesion and Enhances Experimental Metastasis of Murine Mammary Carcinoma. Clin. Exp. Metastasis 1995, 13, 16–22. [Google Scholar] [CrossRef]
- Pelicano, H.; Lu, W.; Zhou, Y.; Zhang, W.; Chen, Z.; Hu, Y.; Huang, P. Mitochondrial Dysfunction and Reactive Oxygen Species Imbalance Promote Breast Cancer Cell Motility through a CXCL14-Mediated Mechanism. Cancer Res. 2009, 69, 2375–2383. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haikazian, S.; Olson, M.F. MICAL1 Monooxygenase in Autosomal Dominant Lateral Temporal Epilepsy: Role in Cytoskeletal Regulation and Relation to Cancer. Genes 2022, 13, 715. https://doi.org/10.3390/genes13050715
Haikazian S, Olson MF. MICAL1 Monooxygenase in Autosomal Dominant Lateral Temporal Epilepsy: Role in Cytoskeletal Regulation and Relation to Cancer. Genes. 2022; 13(5):715. https://doi.org/10.3390/genes13050715
Chicago/Turabian StyleHaikazian, Sipan, and Michael F. Olson. 2022. "MICAL1 Monooxygenase in Autosomal Dominant Lateral Temporal Epilepsy: Role in Cytoskeletal Regulation and Relation to Cancer" Genes 13, no. 5: 715. https://doi.org/10.3390/genes13050715
APA StyleHaikazian, S., & Olson, M. F. (2022). MICAL1 Monooxygenase in Autosomal Dominant Lateral Temporal Epilepsy: Role in Cytoskeletal Regulation and Relation to Cancer. Genes, 13(5), 715. https://doi.org/10.3390/genes13050715