
A Risk Model of Eight Immune-Related Genes Predicting Prognostic Response to Immune Therapies for Gastric Cancer



Abstract
:1. Introduction
2. Methods
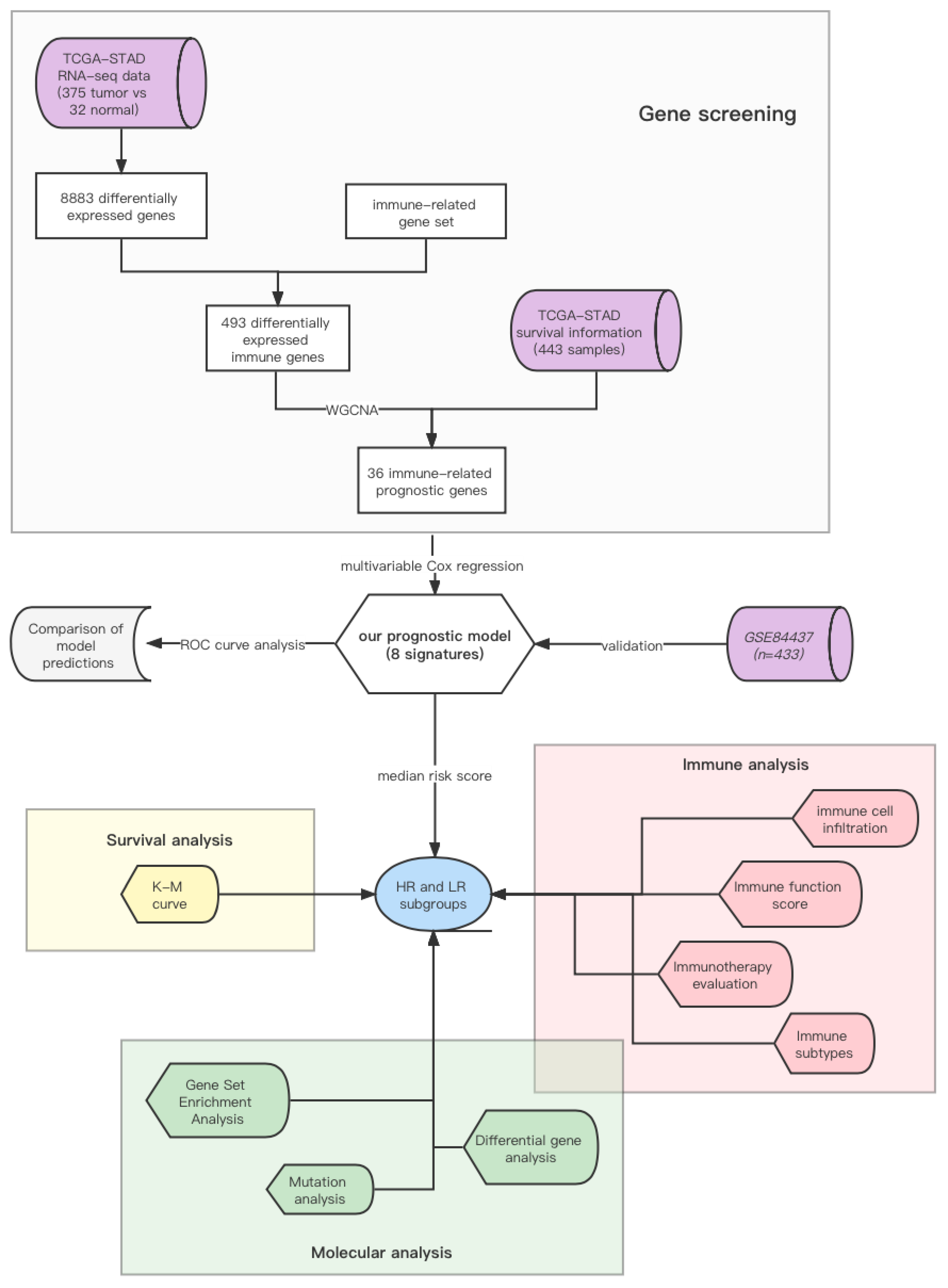
2.1. Data Collection and Processing
2.2. Identification of Prognostic Immune-Related Genes
2.3. Construction and Validation of the Risk-Score Model
2.4. Comprehensive Analysis of Molecular and Immune Characteristics in HR and LR Subgroups
2.5. Verifying the Model Accuracy and Contrasting It with Traditional Ones
2.6. Statistical Analysis
3. Results
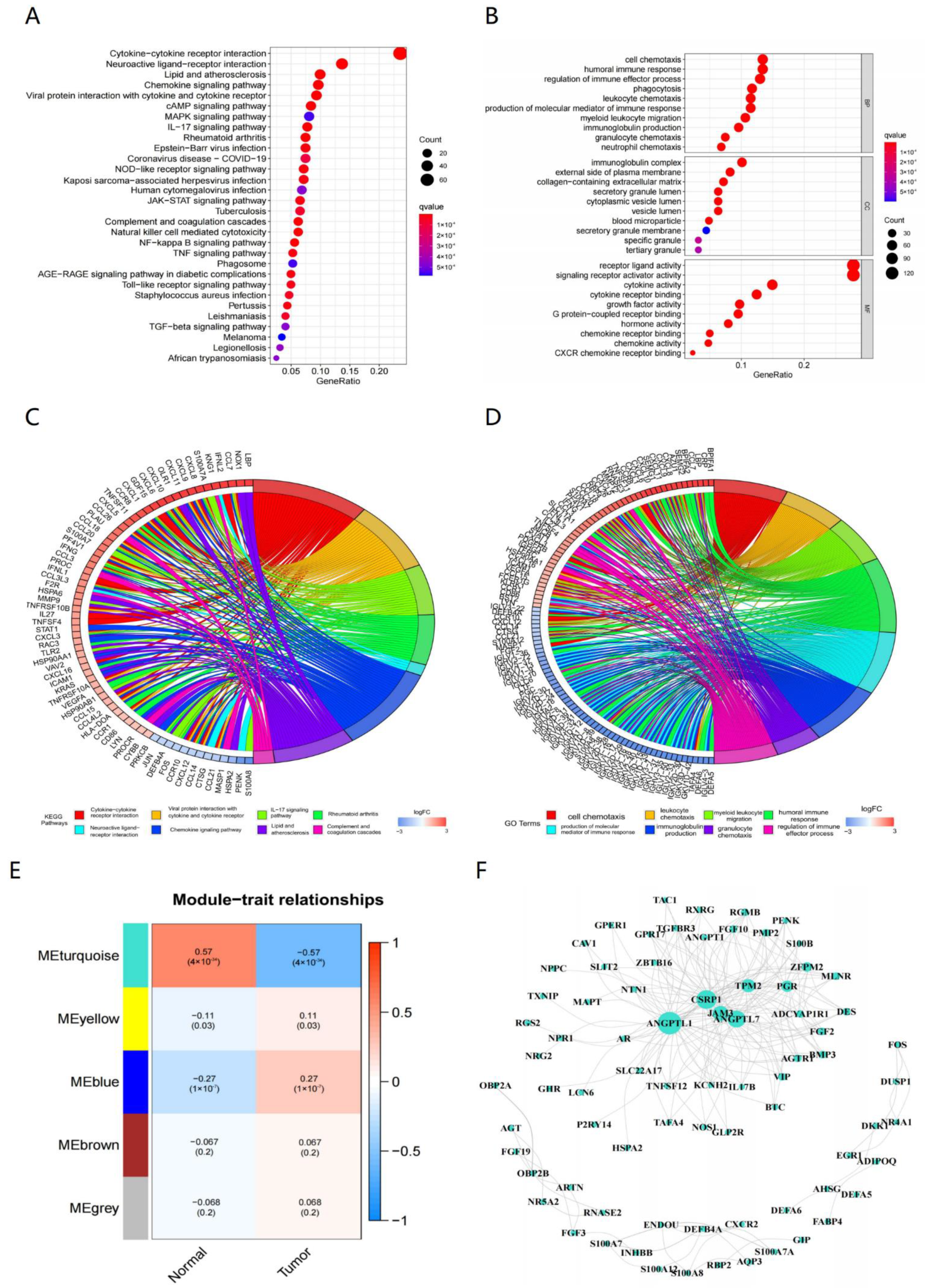
3.1. Outcomes of Prognostic Immune-Related Genes
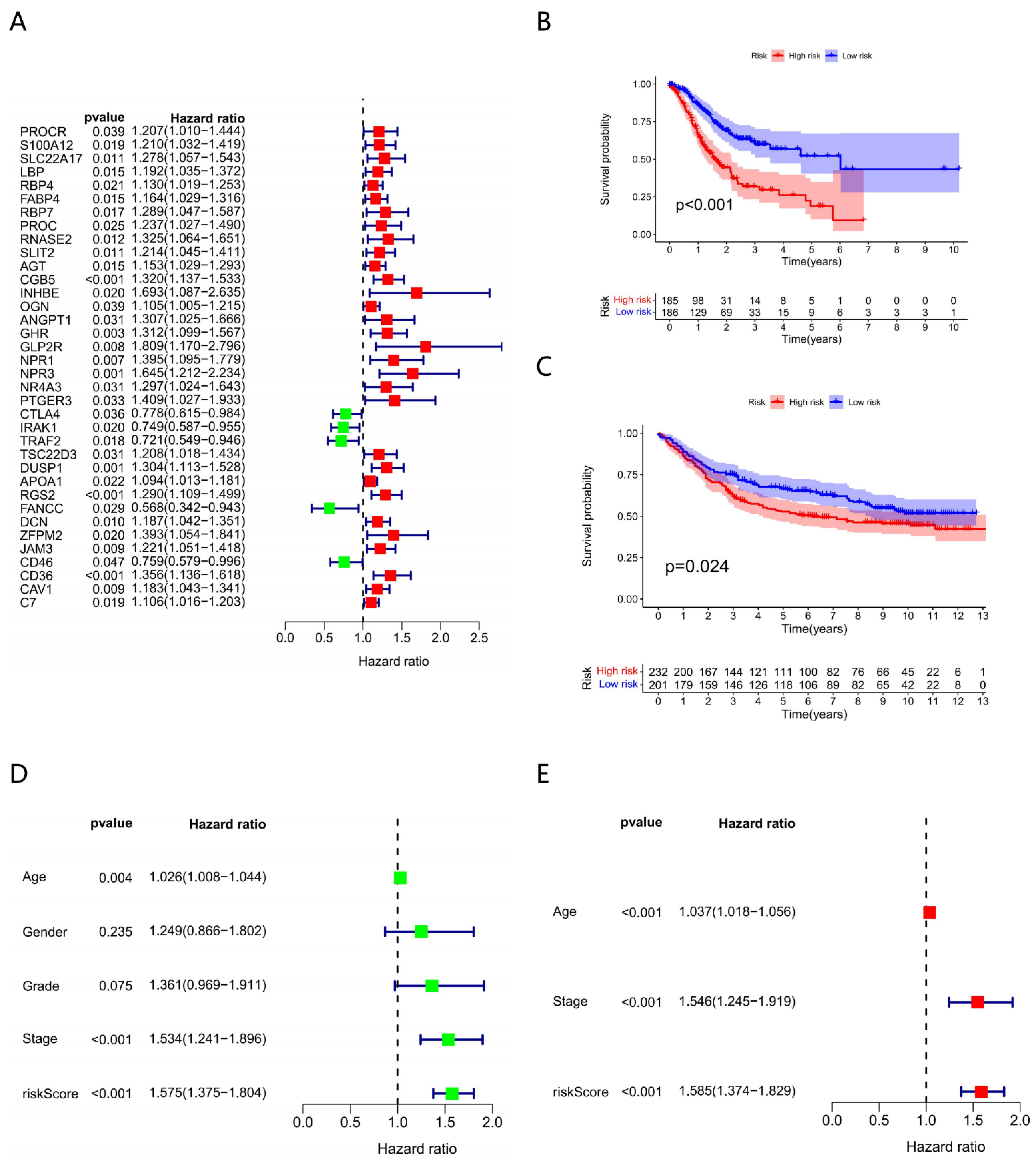
3.2. Constructing Model via 8 Biomarkers
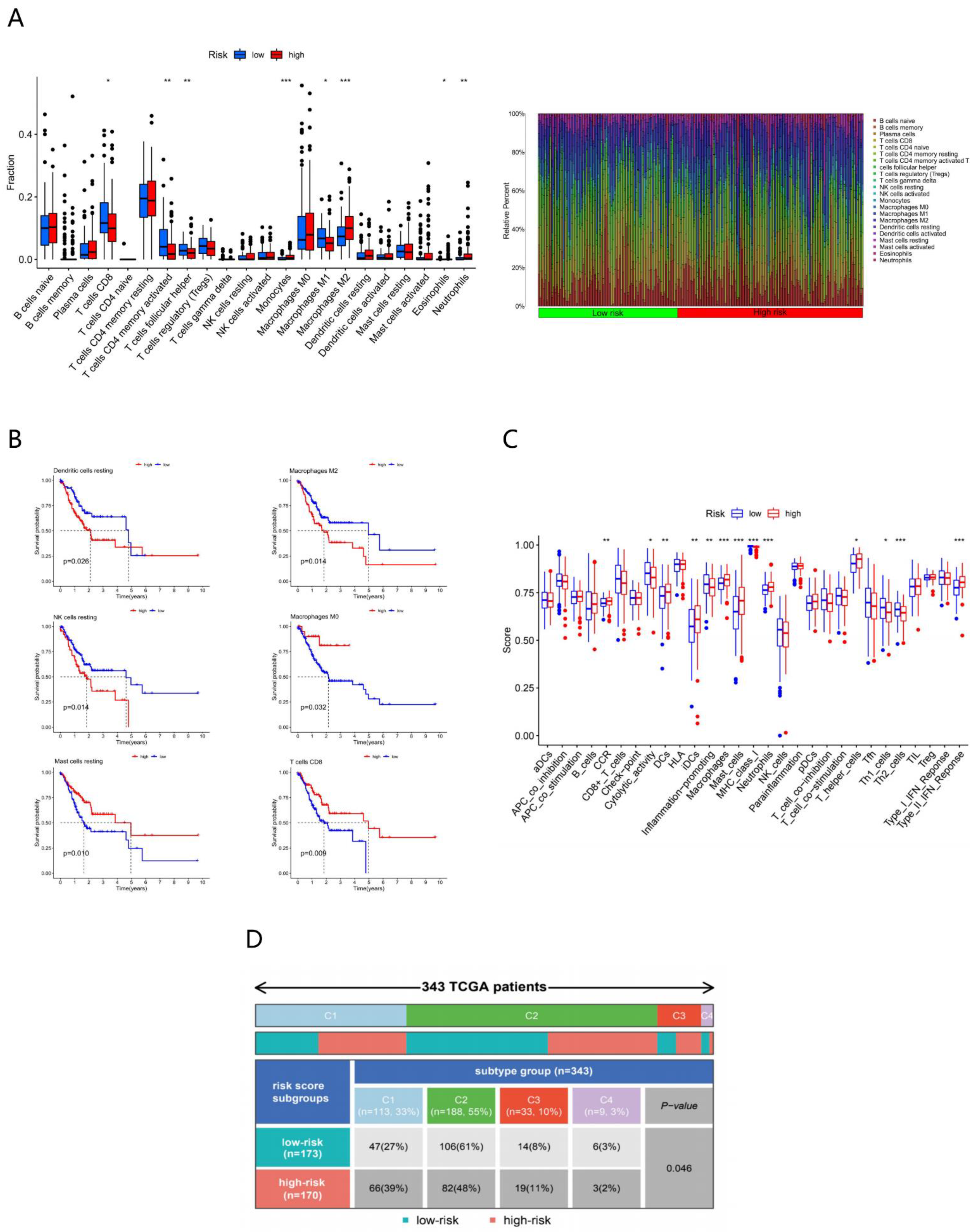
3.3. Clinical Characteristics of Different Risk Subgroups
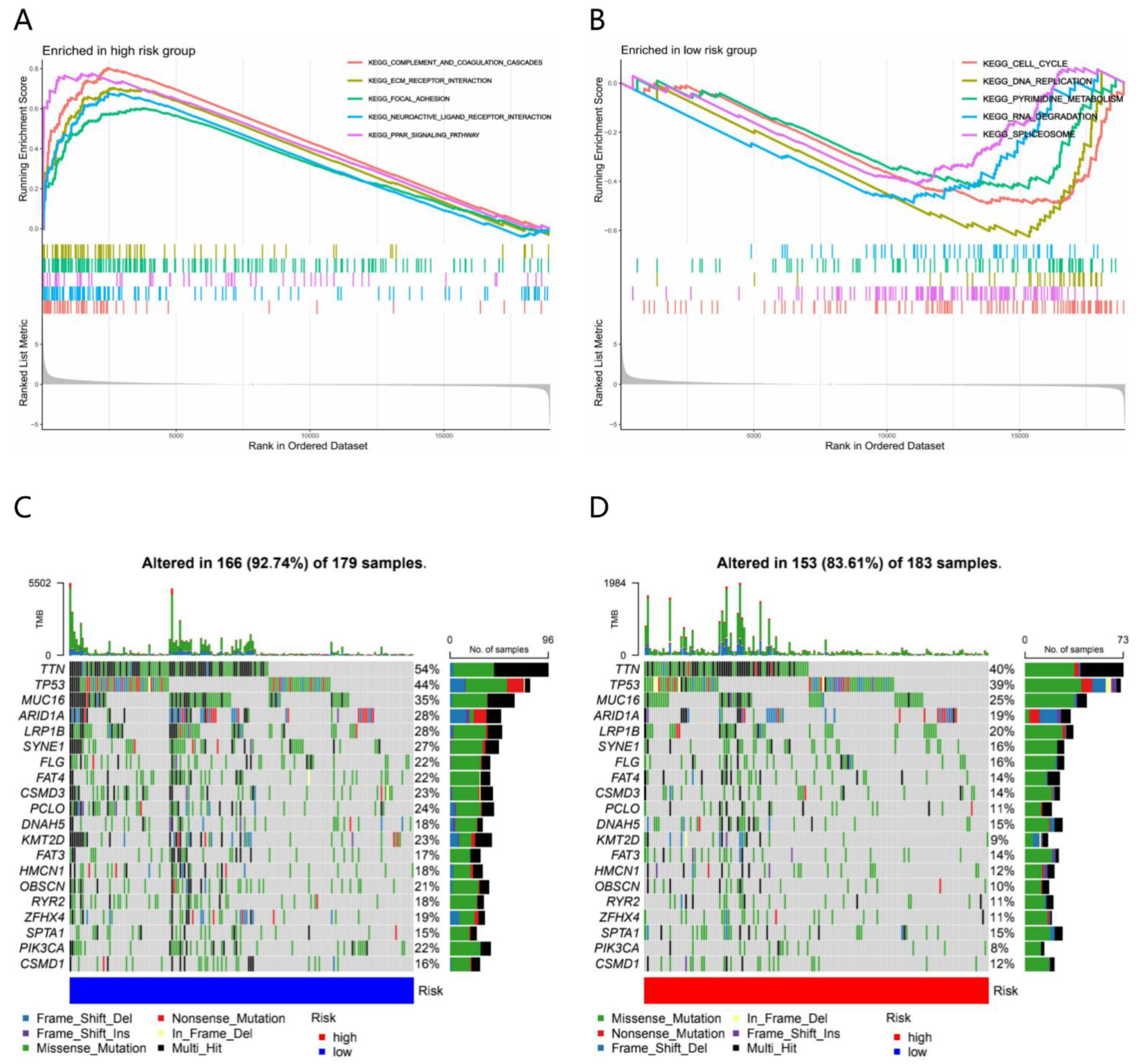
3.4. Molecular Characteristics of Different Risk Subgroups
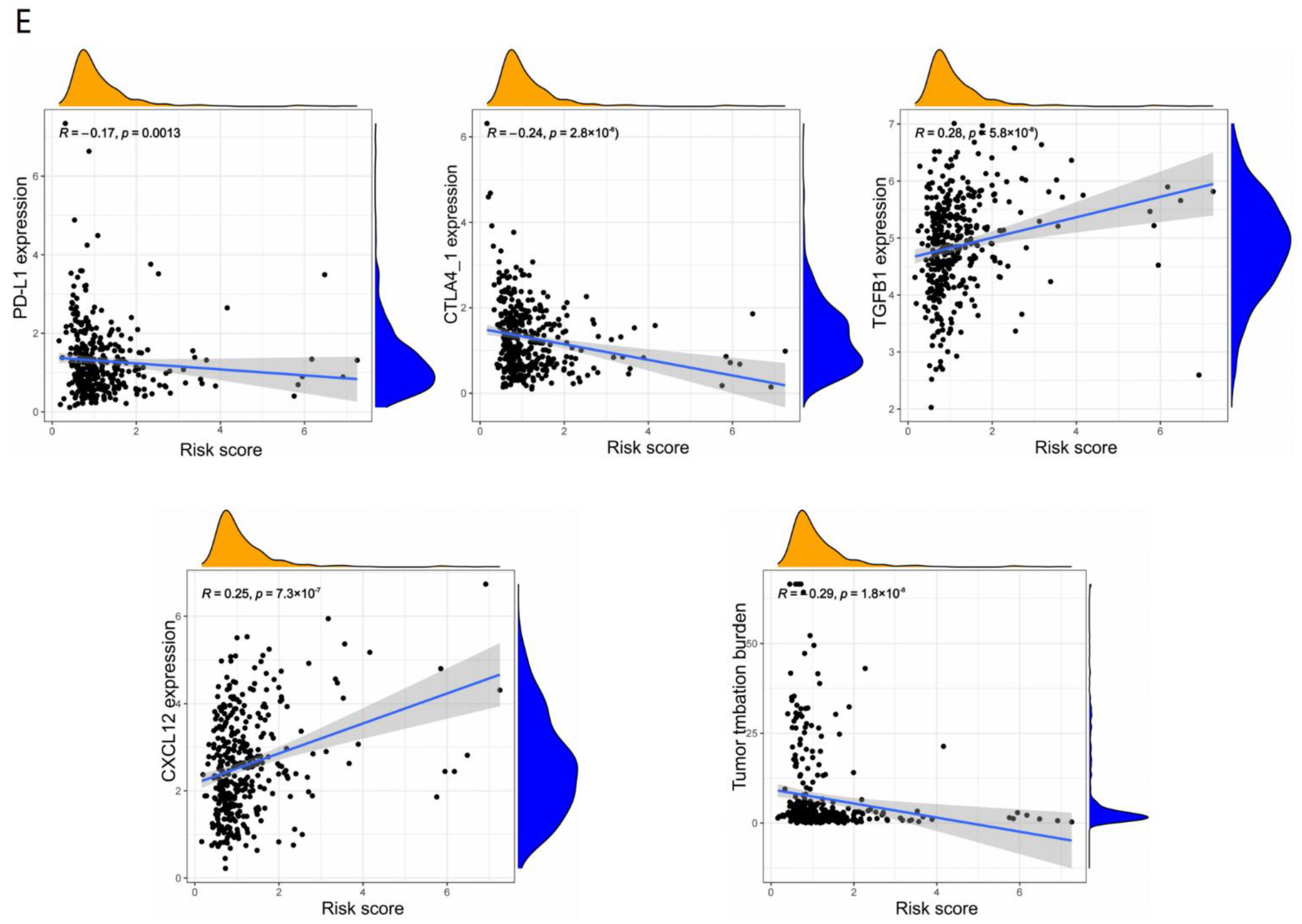
3.5. Immune Characteristics of Different Risk Subgroups
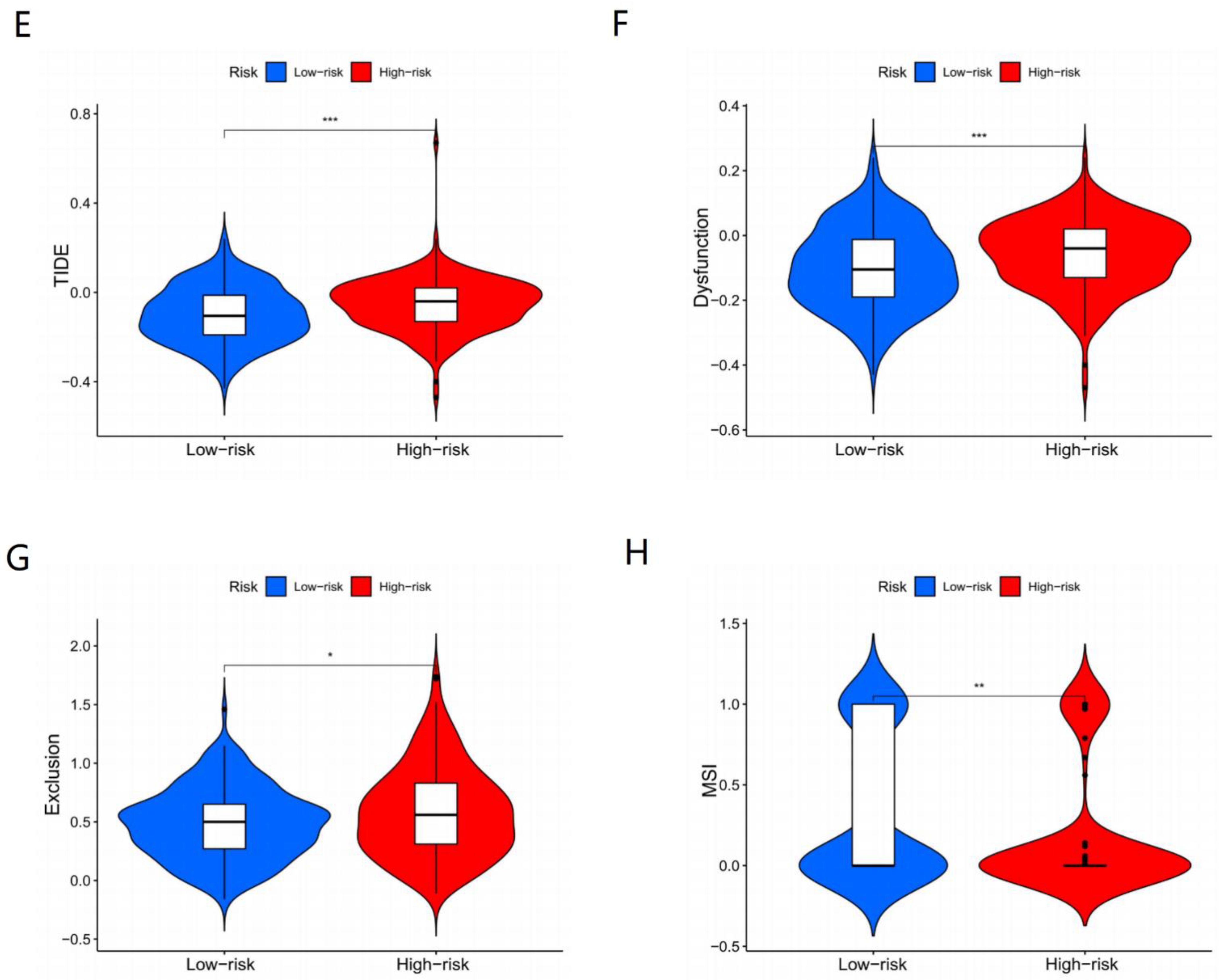
3.6. Immunotherapy Effects of Different Subgroups
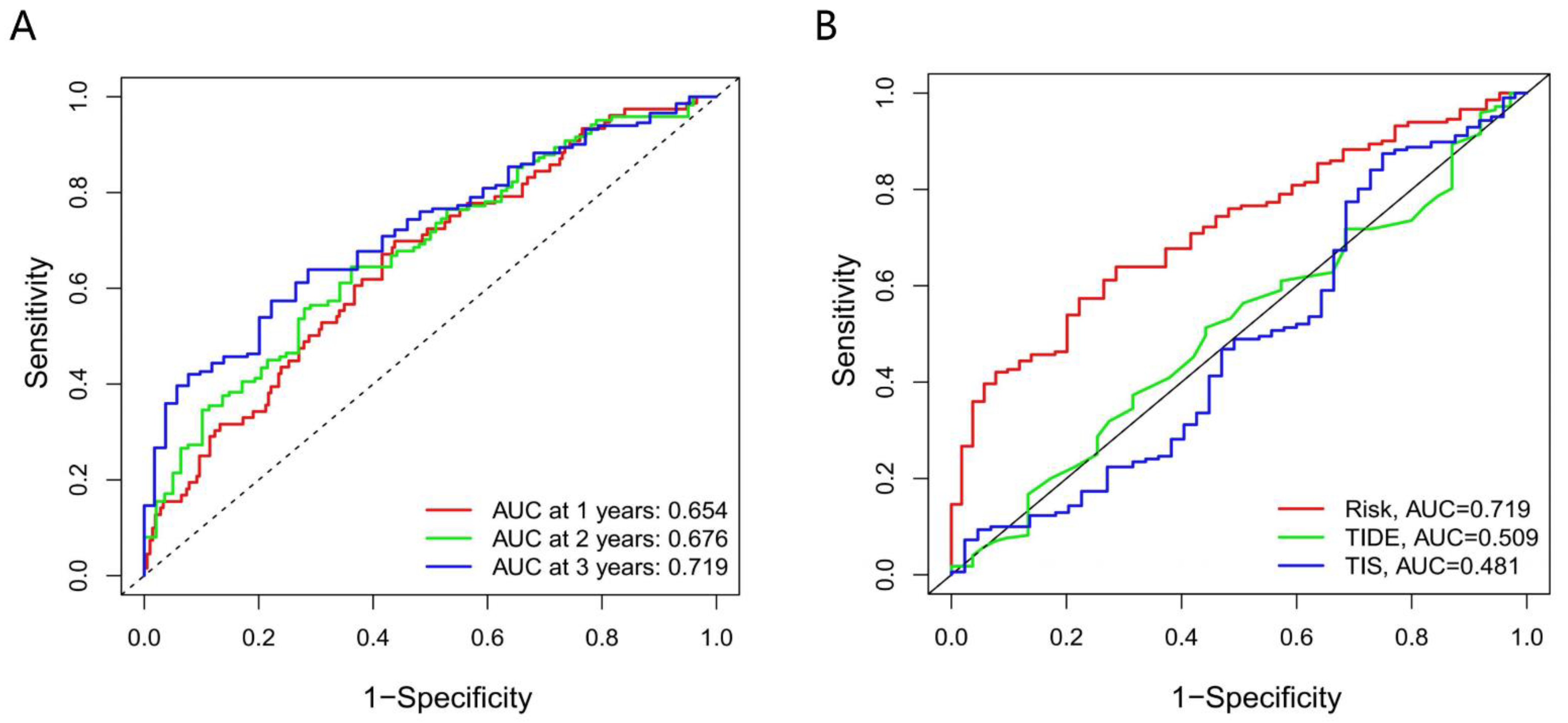
3.7. The Accuracy of the Prognostic Model
4. Discussion
4.1. CTLA4 Plays a Vital Role in Response to ICI
4.2. Relationship between Immune-Related Genes and Cancers
4.3. Somatic Mutation, TMB and TME Corelated with Response to ICI Treatment
4.4. Relationship between Immune Subtypes of Solid Tumors and Our Model
4.5. MSI and TIDE Support Our Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.-K.; Boku, N.; Satoh, T.; Ryu, M.-H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.-S.; Muro, K.; Kang, W.K.; et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 2461–2471. [Google Scholar] [CrossRef]
- Moehler, M.; Ryu, M.-H.; Dvorkin, M.; Lee, K.-W.; Coşkun, H.; Wong, R.; Chung, H.; Poltoratsky, A.; Tsuji, A.; Yen, C.J.; et al. Maintenance avelumab versus continuation of first-line chemotherapy in gastric cancer: JAVELIN Gastric 100 study design. Futur. Oncol. 2019, 15, 567–577. [Google Scholar] [CrossRef]
- Kono, K.; Nakajima, S.; Mimura, K. Current status of immune checkpoint inhibitors for gastric cancer. Gastric Cancer 2020, 23, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Jing, C.; Wang, J.; Zhu, M.; Bai, Z.; Zhao, B.; Zhang, J.; Yin, J.; Yang, X.; Liu, Z.; Zhang, Z.; et al. Camrelizumab combined with apatinib and S-1 as second-line treatment for patients with advanced gastric or gastroesophageal junction adenocarcinoma: A phase 2, single-arm, prospective study. Cancer Immunol. Immunother. 2022, 1–12. [Google Scholar] [CrossRef]
- Aggarwal, C.; Prawira, A.; Antonia, S.; Rahma, O.; Tolcher, A.; Cohen, R.B.; Lou, Y.; Hauke, R.; Vogelzang, N.; Zandberg, D.P.; et al. Dual checkpoint targeting of B7-H3 and PD-1 with enoblituzumab and pembrolizumab in advanced solid tumors: Interim results from a multicenter phase I/II trial. J. Immunother. Cancer 2022, 10, e004424. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients with Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Cho, J.Y.; Kim, Y.H.; Kim, J.W.; Di Bartolomeo, M.; Ajani, J.A.; Yamaguchi, K.; Balogh, A.; Sanchez, T.; Moehler, M. Efficacy of Sequential Ipilimumab Monotherapy versus Best Supportive Care for Unresectable Locally Advanced/Metastatic Gastric or Gastroesophageal Junction Cancer. Clin. Cancer Res. 2017, 23, 5671–5678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pento, J.T. Monoclonal Antibodies for the Treatment of Cancer. Anticancer Res. 2017, 37, 5935–5939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, E.; Thuss-Patience, P.C. Immune Checkpoint Inhibition in Gastro-Oesophageal Cancer. Oncol. Res. Treat. 2018, 41, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef] [Green Version]
- Davalieva, K.; Kiprijanovska, S.; Kostovska, I.M.; Stavridis, S.; Stankov, O.; Komina, S.; Petrusevska, G.; Polenakovic, M. Comparative Proteomics Analysis of Urine Reveals Down-Regulation of Acute Phase Response Signaling and LXR/RXR Activation Pathways in Prostate Cancer. Proteomes 2017, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Szczerba, A.; Śliwa, A.; Kubiczak, M.; Nowak-Markwitz, E.; Jankowska, A. Human chorionic gonadotropin β subunit affects the expression of apoptosis-regulating factors in ovarian cancer. Oncol. Rep. 2016, 35, 538–545. [Google Scholar] [CrossRef]
- Tsampalas, M.; Gridelet, V.; Berndt, S.; Foidart, J.-M.; Geenen, V.; D’Hauterive, S.P. Human chorionic gonadotropin: A hormone with immunological and angiogenic properties. J. Reprod. Immunol. 2010, 85, 93–98. [Google Scholar] [CrossRef]
- Li, Y.; He, X.; Fan, L.; Zhang, X.; Xu, Y.; Xu, X. Identification of a novel immune prognostic model in gastric cancer. Clin. Transl. Oncol. 2020, 23, 846–855. [Google Scholar] [CrossRef]
- Wang, L.; Gu, W.; Ni, H. Construction of a prognostic value model in papillary renal cell carcinoma by immune-related genes. Medicine 2021, 100, e24903. [Google Scholar] [CrossRef]
- Brüning, A.; Matsingou, C.; Brem, G.J.; Rahmeh, M.; Mylonas, I. Inhibin β E is upregulated by drug-induced endoplasmic reticulum stress as a transcriptional target gene of ATF4. Toxicol. Appl. Pharmacol. 2012, 264, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Shi, H.; Gatzke, F.; Bennett, A.M. Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cell Mol. Life Sci. 2012, 70, 223–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Wang, Y.; Shi, L.Z.; Kanneganti, T.-D.; Chi, H. Signaling by the Phosphatase MKP-1 in Dendritic Cells Imprints Distinct Effector and Regulatory T Cell Fates. Immunity 2011, 35, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.R.; Villanueva, A.I.; Read, L.R.; Brisbin, J.T.; Bhaumik, S.K.; Murali-Krishna, K.; Sharif, S.; Lamarre, J. CpG oligonucleotide-mediated co-stimulation of mouse invariant natural killer T cells negatively regulates their activation status. Cell Tissue Res. 2017, 369, 541–554. [Google Scholar] [CrossRef]
- CD36 Activity Causes Ferroptosis in Tumor-Infiltrating CD8(+) T Cells. Cancer Discov. 2021, 11, OF24. [CrossRef]
- Xu, S.; Chaudhary, O.; Rodríguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity 2021, 54, 1561–1577.e7. [Google Scholar] [CrossRef]
- Su, C.; Wang, X.; Zhou, J.; Zhao, J.; Zhou, F.; Zhao, G.; Xu, X.; Zou, X.; Zhu, B.; Jia, Q. Titin mutation in circulatory tumor DNA is associated with efficacy to immune checkpoint blockade in advanced non-small cell lung cancer. Transl. Lung Cancer Res. 2021, 10, 1256–1265. [Google Scholar] [CrossRef]
- Li, L.; Li, M.; Wang, X. Cancer type-dependent correlations between TP53 mutations and antitumor immunity. DNA Repair 2020, 88, 102785. [Google Scholar] [CrossRef]
- Nie, K.; Zheng, Z.; Wen, Y.; Shi, L.; Xu, S.; Wang, X.; Zhou, Y.; Fu, B.; Li, X.; Deng, Z.; et al. Construction and validation of a TP53-associated immune prognostic model for gastric cancer. Genomics 2020, 112, 4788–4795. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Bi, F.; Chen, Y.; Yang, Q. Significance of tumor mutation burden combined with immune infiltrates in the progression and prognosis of ovarian cancer. Cancer Cell Int. 2020, 20, 373. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liang, X.; Wang, Y.; Cheng, A.; Zhang, H.; Qin, C.; Wang, Z. Significance of Tumor Mutation Burden Combined with Immune Infiltrates in the Progression and Prognosis of Advanced Gastric Cancer. Front. Genet. 2021, 12, 642608. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Li, Z.; Suo, B.; Long, G.; Gao, Y.; Song, J.; Zhang, M.; Feng, B.; Shang, C.; Wang, D. Exosomal miRNA-16-5p Derived From M1 Macrophages Enhances T Cell-Dependent Immune Response by Regulating PD-L1 in Gastric Cancer. Front. Cell Dev. Biol. 2020, 8, 572689. [Google Scholar] [CrossRef]
- Chen, Y.-P.; Wang, Y.-Q.; Lv, J.-W.; Li, Y.-Q.; Chua, M.L.K.; Le, Q.-T.; Lee, N.; Colevas, A.D.; Seiwert, T.; Hayes, D.N.; et al. Identification and validation of novel microenvironment-based immune molecular subgroups of head and neck squamous cell carcinoma: Implications for immunotherapy. Ann. Oncol. 2019, 30, 68–75. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source_Reference_ID | Gene Name | Coefficient |
|---|---|---|
| NM_00293.4 | RNASE2 | 0.276627343597117 |
| NM_033043.1 | CGB5 | 0.255445846089108 |
| NM_031479.3 | INHBE | 0.501097892816982 |
| NM_003584.1 | DUSP1 | 0.259804690773917 |
| NM_000039.1 | APOA1 | 0.07098152433948 |
| NM_001001548.1 | CD36 | 0.234091941360161 |
| NM_198712.2 | PTGER3 | −0.348295793637423 |
| NM_005214.3 | CTLA4 | −0.341191391278679 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, M.; Zhang, Y.; Mao, R.; Zhu, C.; Zhao, R.; Jin, L. A Risk Model of Eight Immune-Related Genes Predicting Prognostic Response to Immune Therapies for Gastric Cancer. Genes 2022, 13, 720. https://doi.org/10.3390/genes13050720
Yu M, Zhang Y, Mao R, Zhu C, Zhao R, Jin L. A Risk Model of Eight Immune-Related Genes Predicting Prognostic Response to Immune Therapies for Gastric Cancer. Genes. 2022; 13(5):720. https://doi.org/10.3390/genes13050720
Chicago/Turabian StyleYu, Miao, Yi Zhang, Rongchen Mao, Chao Zhu, Ruixue Zhao, and Lai Jin. 2022. "A Risk Model of Eight Immune-Related Genes Predicting Prognostic Response to Immune Therapies for Gastric Cancer" Genes 13, no. 5: 720. https://doi.org/10.3390/genes13050720
APA StyleYu, M., Zhang, Y., Mao, R., Zhu, C., Zhao, R., & Jin, L. (2022). A Risk Model of Eight Immune-Related Genes Predicting Prognostic Response to Immune Therapies for Gastric Cancer. Genes, 13(5), 720. https://doi.org/10.3390/genes13050720