
Characterization, Selection, and Trans-Species Polymorphism in the MHC Class II of Heermann’s Gull (Charadriiformes)

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction, and PCR Amplification of MHCIIB
2.2. Cloning, Sequencing, and the Validation of Alleles
2.3. Intraspecific Relationships and the Characterization of MHCIIB Polymorphisms
2.4. Analysis of Selection Signatures in Heermann’s Gull and Other Charadriiformes
2.5. Historical Demographic Changes in Heermann’s Gulls
2.6. Interspecific Phylogenetic Relationships of MHCIIB in Larids and Other Charadriiformes
3. Results
3.1. MHCIIB Recovered by Cloning in Heermann’s Gull
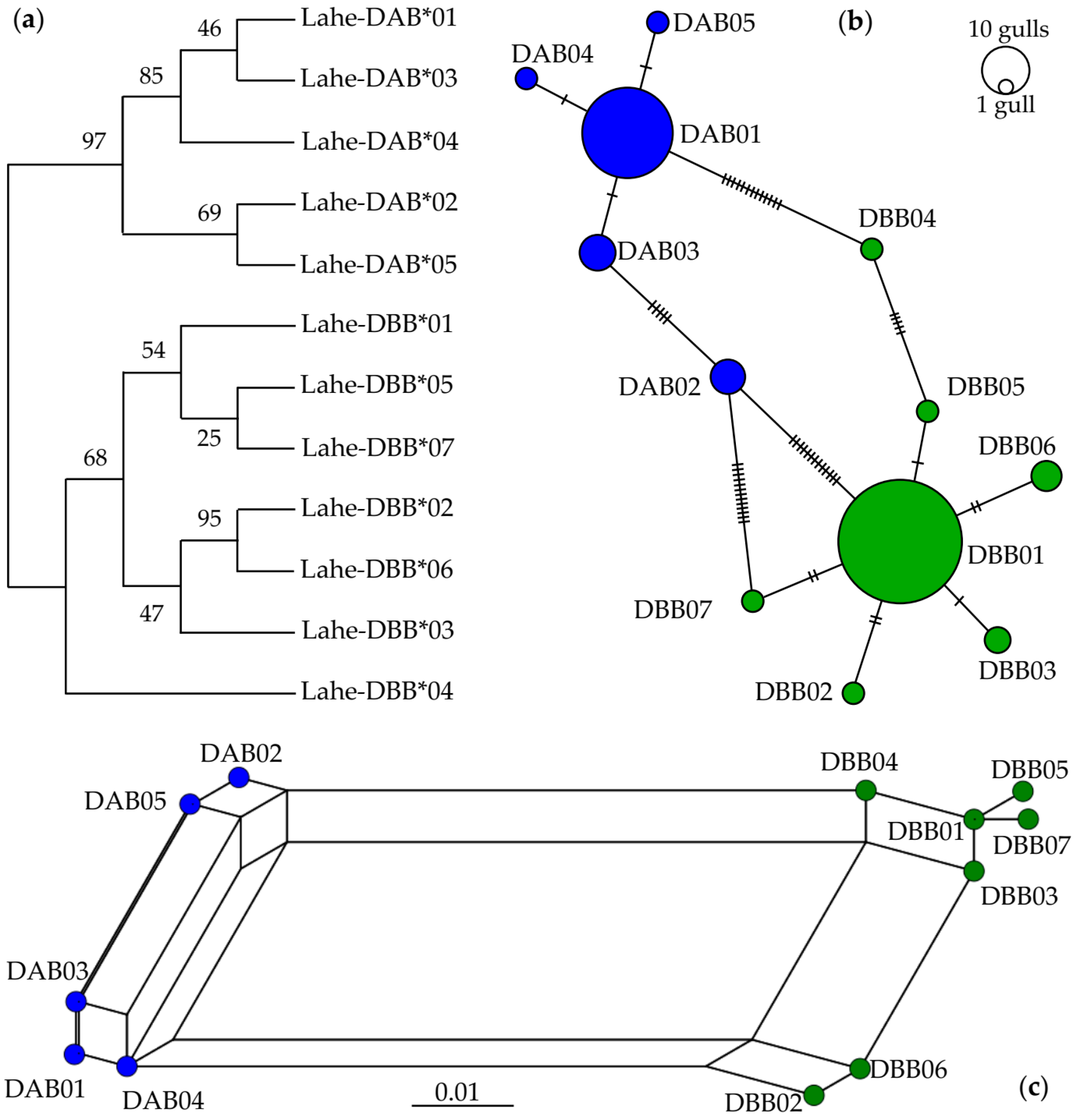
3.2. Inferred Duplication by Phylogenetic Relationships within Heermann’s Gull MHCIIB
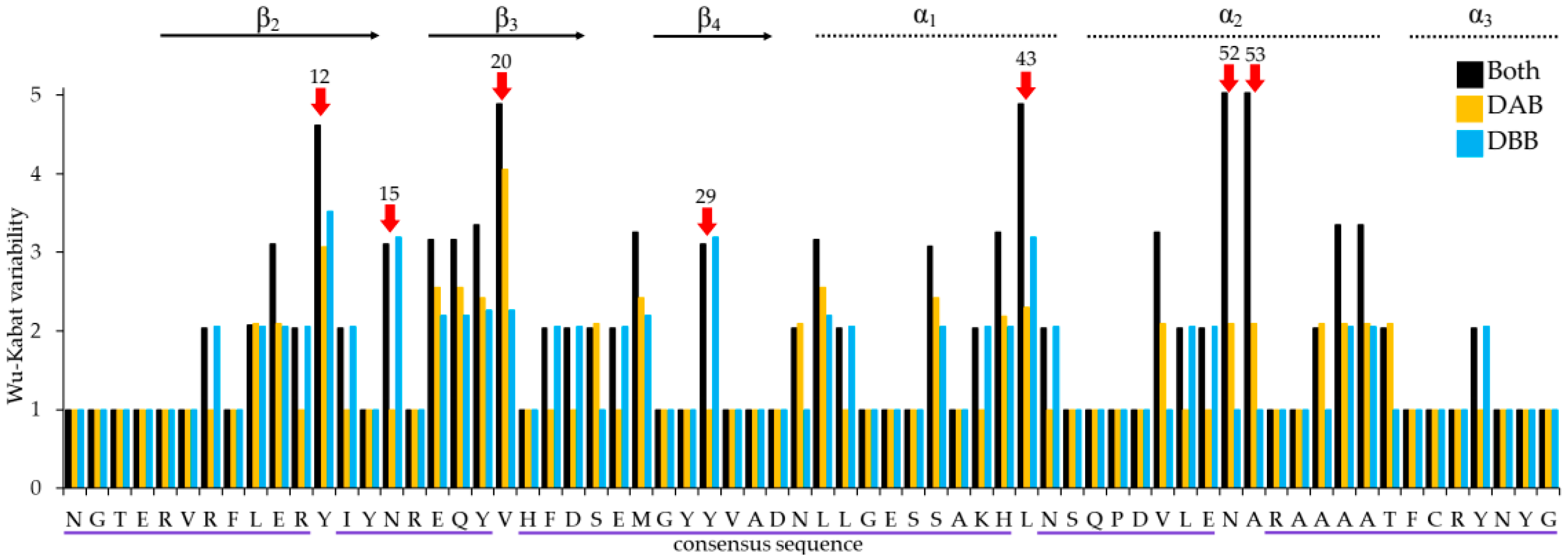
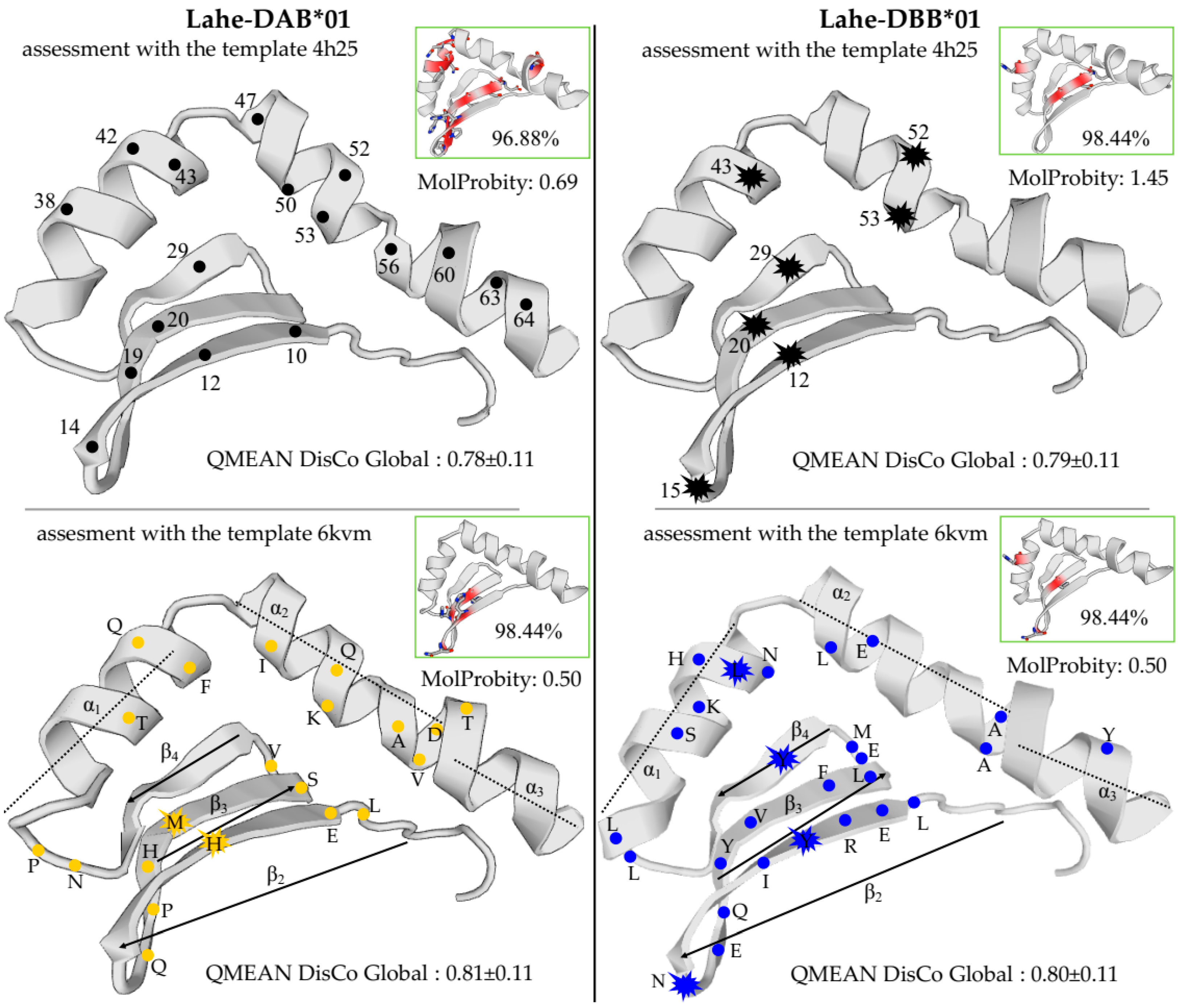
3.3. Characterization Heermann’s Gull MHCIIB Polymorphism
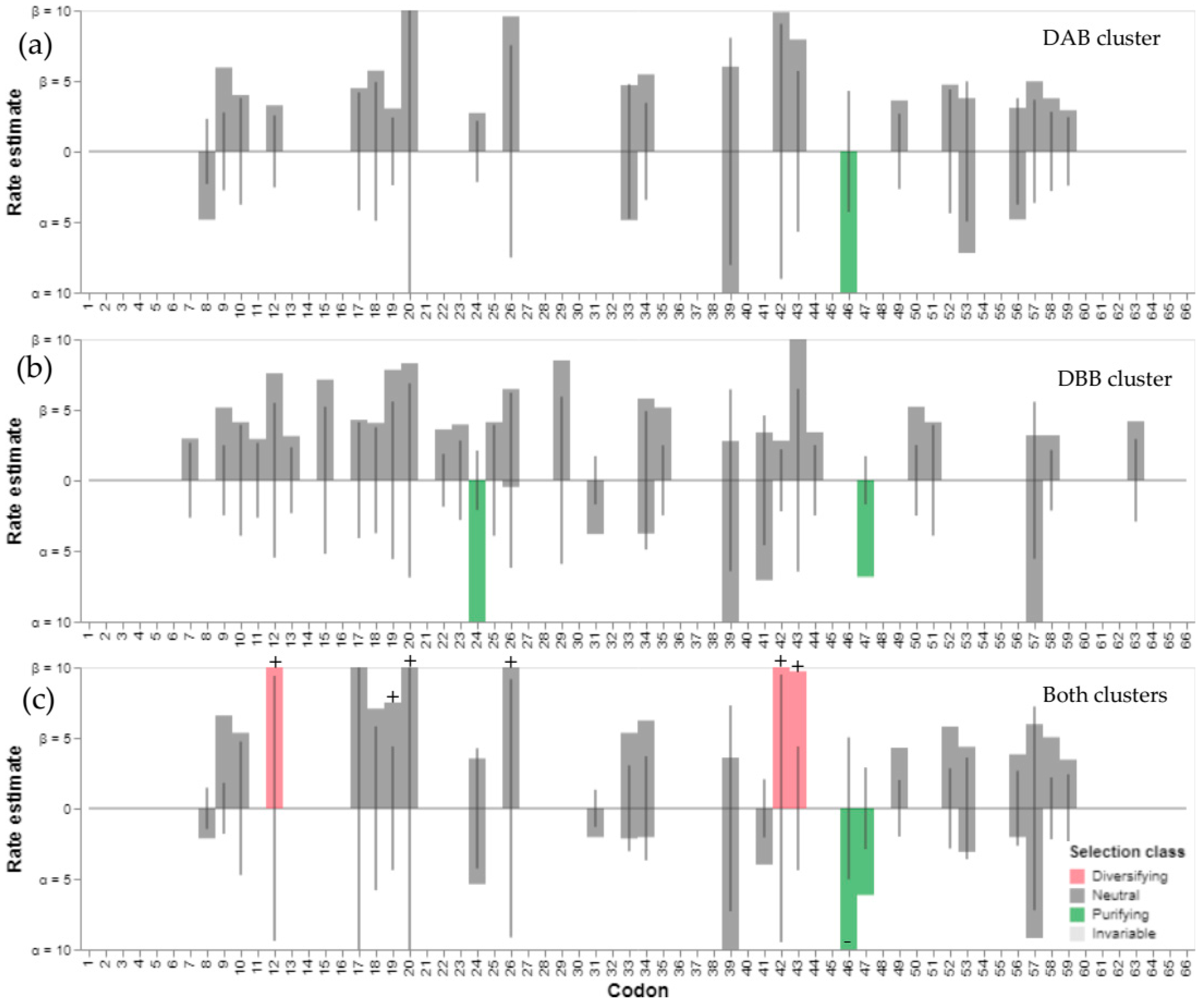
3.4. Analysis of Selection
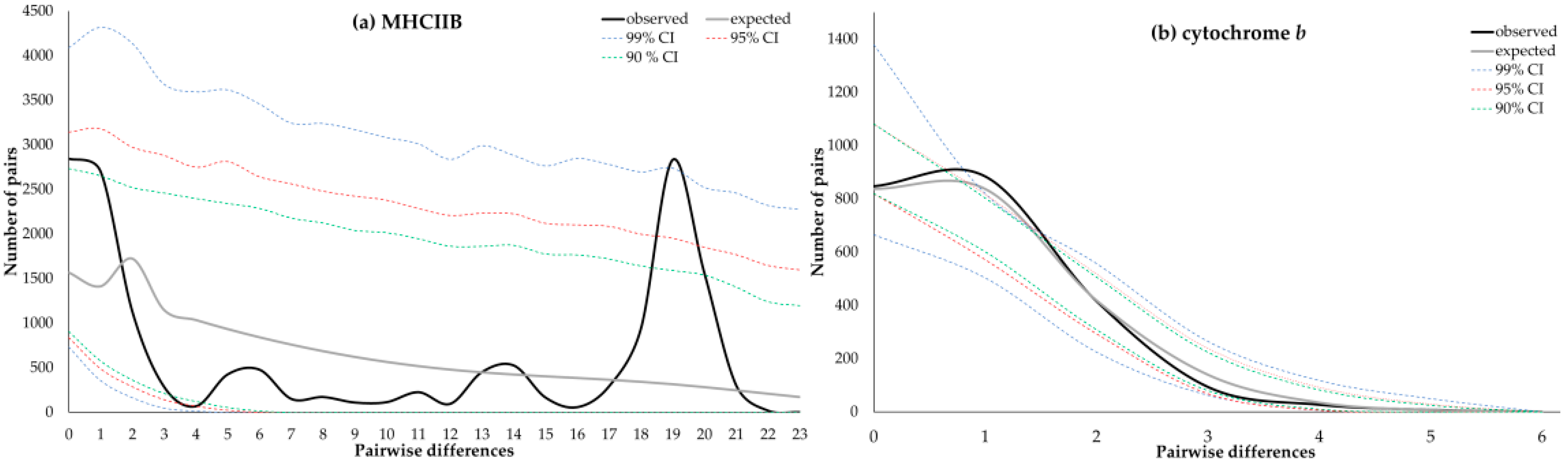
3.5. Demographic Expansion in Heermann’s Gull from Isla Rasa
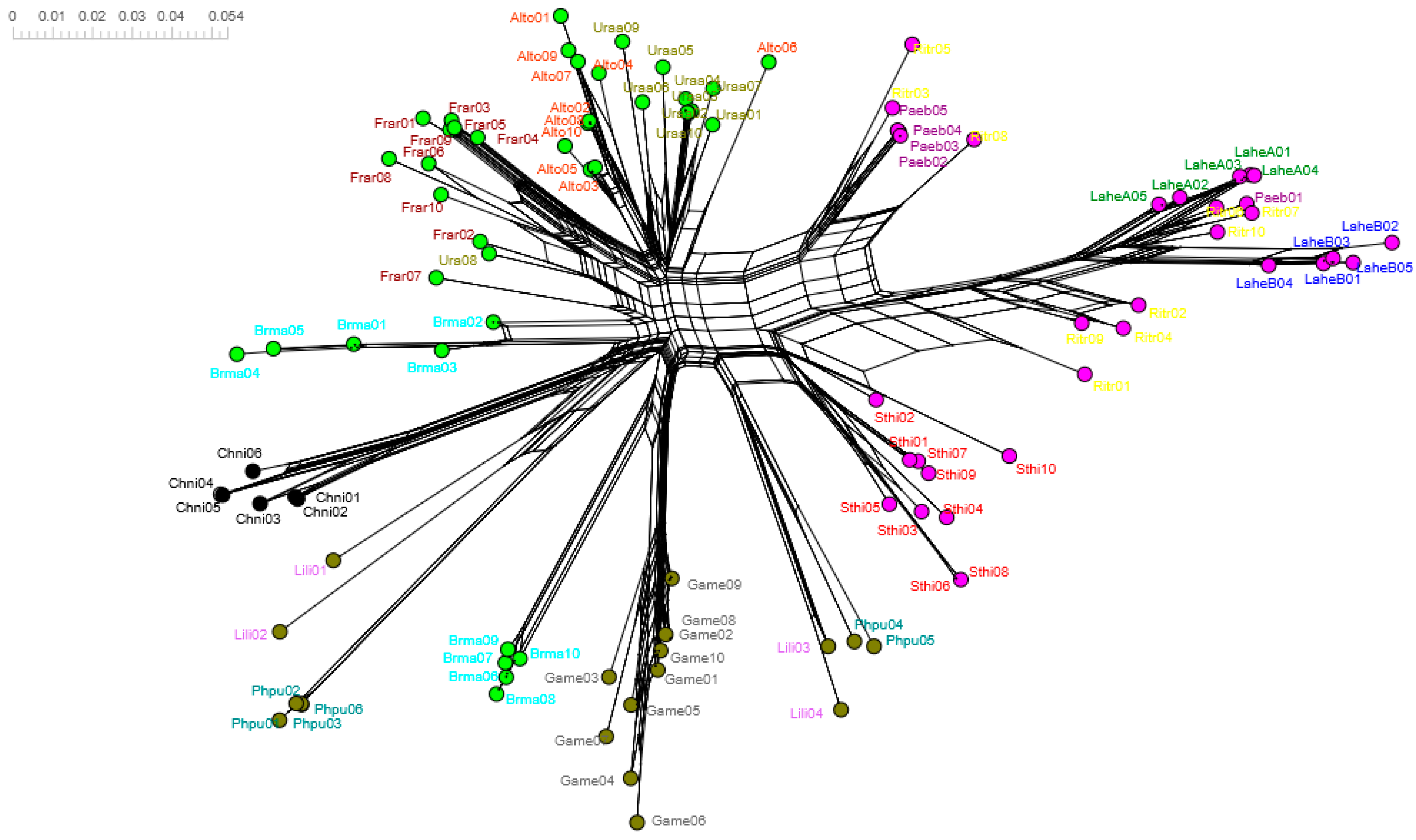
3.6. Trans-Species Polymorphism in Charadriiformes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, S.V.; Hedrick, P.W. Evolution and ecology of MHC molecules: From genomics to sexual selection. Trends Ecol. Evol. 1998, 13, 305–311. [Google Scholar] [CrossRef]
- Jurewicz, M.M.; Stern, L.J. Class II MHC antigen processing in immune tolerance and inflammation. Immunogenetics 2019, 71, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Bentkowski, P.; Radwan, J. Evolution of major histocompatibility complex gene copy number. PLoS Comput. Biol. 2019, 15, e1007015. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.C.; Lambert, D.M. Gene duplication and gene conversion in class II MHC genes of New Zealand robins (Petroicidae). Immunogenetics 2004, 56, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Sato, A.; Nagl, S.; O’hUigín, C. Molecular trans-species polymorphism. Annu. Rev. Ecol. Syst. 1998, 29, 1–21. [Google Scholar] [CrossRef]
- Vlček, J.; Hoeck, P.E.A.; Keller, L.F.; Wayhart, J.P.; Dolinová, I.; Štefka, J. Balancing selection and genetic drift create unusual patterns of MHCIIβ variation in Galápagos mockingbirds. Mol. Ecol. 2016, 25, 4757–4772. [Google Scholar] [CrossRef]
- Kamiya, T.; O’Dwyer, K.; Westerdahl, H.; Senior, A.; Nakagawa, S. A quantitative review of MHC-based mating preference: The role of diversity and dissimilarity. Mol. Ecol. 2014, 23, 5151–5163. [Google Scholar] [CrossRef]
- O’Connor, E.A.; Hasselquist, D.; Nilsson, J.Å.; Westerdahl, H.; Cornwallis, C.K. Wetter climates select for higher immune gene diversity in resident, but not migratory, songbirds. Proc. R. Soc. B Biol. Sci. 2020, 287, 20192675. [Google Scholar] [CrossRef]
- Arauco-Shapiro, G.; Schumacher, K.I.; Boersma, D.; Bouzat, J.L. The role of demographic history and selection in shaping genetic diversity of the Galápagos penguin (Spheniscus mendiculus). PLoS ONE 2020, 15, e0226439. [Google Scholar] [CrossRef] [Green Version]
- Jamie, G.A.; Meier, J.I. The Persistence of Polymorphisms across Species Radiations. Trends Ecol. Evol. 2020, 35, 795–808. [Google Scholar] [CrossRef]
- Radwan, J.; Babik, W.; Kaufman, J.; Lenz, T.L.; Winternitz, J. Advances in the Evolutionary Understanding of MHC Polymorphism. Trends Genet. 2020, 36, 298–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, J.; Salomonsen, J.; Flajnik, M. Evolutionary conservation of MHC class I and class II molecules--different yet the same. Semin. Immunol. 1994, 6, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Strandh, M.; Westerdahl, H.; Pontarp, M.; Canbäck, B.; Dubois, M.P.; Miquel, C.; Taberlet, P.; Bonadonna, F. Major histocompatibility complex class II compatibility, but not class I, predicts mate choice in a bird with highly developed olfaction. Proc. R. Soc. B Biol. Sci. 2012, 279, 4457–4463. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Song, M.; Lai, J.; Sun, J.; Gong, Q. Molecular polymorphism and expression of MHC I α, II α, II β and II invariant chain in the critically endangered Dabry’s sturgeon (Acipenser dabryanus). Dev. Comp. Immunol. 2020, 103, 103494. [Google Scholar] [CrossRef]
- Gösser, F.; Schartl, M.; García-De León, F.J.; Tollrian, R.; Lampert, K.P. Red Queen revisited: Immune gene diversity and parasite load in the asexual Poecilia formosa versus its sexual host species P. mexicana. PLoS ONE 2019, 14, e0219000. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, E.A.; Cornwallis, C.K.; Hasselquist, D.; Nilsson, J.Å.; Westerdahl, H. The evolution of immunity in relation to colonization and migration. Nat. Ecol. Evol. 2018, 2, 841–849. [Google Scholar] [CrossRef]
- Hedrick, P.W. Pathogen resistance and genetic variation at MHC loci. Evolution 2002, 56, 1902–1908. [Google Scholar] [CrossRef]
- Antonides, J.; Mathur, S.; DeWoody, J.A. Episodic positive diversifying selection on key immune system genes in major avian lineages. Genetica 2019, 147, 337–350. [Google Scholar] [CrossRef]
- Slade, J.W.G.; Watson, M.J.; MacDougall-Shackleton, E.A. “Balancing” balancing selection? Assortative mating at the major histocompatibility complex despite molecular signatures of balancing selection. Ecol. Evol. 2019, 9, 5146–5157. [Google Scholar] [CrossRef]
- Brisson, D. Negative frequency-dependent selection is frequently confounding. Front. Ecol. Evol. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milinski, M. The Major Histocompatibility Complex, Sexual Selection, and Mate Choice. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 159–186. [Google Scholar] [CrossRef]
- Bertram, J.; Masel, J. Different mechanisms drive the maintenance of polymorphism at loci subject to strong versus weak fluctuating selection. Evolution 2019, 73, 883–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.L.; Nei, M. Maintenance of MHC polymorphism. Nature 1992, 355, 402–403. [Google Scholar] [CrossRef]
- Lenz, T.L.; Hafer, N.; Samonte, I.E.; Yeates, S.E.; Milinski, M. Cryptic haplotype-specific gamete selection yields offspring with optimal MHC immune genes. Evolution 2018, 72, 2478–2490. [Google Scholar] [CrossRef]
- Hess, C.M.; Edwards, S.V. The evolution of the major histocompatibility complex in birds. BioScience 2002, 52, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, J. Antigen processing and presentation: Evolution from a bird’s eye view. Mol. Immunol. 2013, 55, 159–161. [Google Scholar] [CrossRef]
- Kaufman, J.; Milne, S.; Göbel, T.W.F.; Walker, B.A.; Jacob, J.P.; Auffray, C.; Zoorob, R.; Beck, S. The chicken B locus is a minimal essential major histocompatibility complex. Nature 1999, 401, 923–925. [Google Scholar] [CrossRef]
- Miller, M.M.; Taylor, R.L. Brief review of the chicken Major Histocompatibility Complex: The genes, their distribution on chromosome 16, and their contributions to disease resistance. Poult. Sci. 2016, 95, 375–392. [Google Scholar] [CrossRef]
- Goebel, J.; Promerová, M.; Bonadonna, F.; McCoy, K.D.; Serbielle, C.; Strandh, M.; Yannic, G.; Burri, R.; Fumagalli, L. 100 million years of multigene family evolution: Origin and evolution of the avian MHC class IIB. BMC Genom. 2017, 18, 460. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, J. Unfinished Business: Evolution of the MHC and the Adaptive Immune System of Jawed Vertebrates. Annu. Rev. Immunol. 2018, 36, 383–409. [Google Scholar] [CrossRef] [PubMed]
- Burri, R.; Promerová, M.; Goebel, J.; Fumagalli, L. PCR-based isolation of multigene families: Lessons from the avian MHC class IIB. Mol. Ecol. Resour. 2014, 14, 778–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minias, P.; Pikus, E.; Whittingham, L.A.; Dunn, P.O. Evolution of copy number at the MHC varies across the avian tree of life. Genome Biol. Evol. 2019, 11, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Burri, R.; Hirzel, H.N.; Salamin, N.; Roulin, A.; Fumagalli, L. Evolutionary patterns of MHC class II B in owls and their implications for the understanding of avian MHC evolution. Mol. Biol. Evol. 2008, 25, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Minias, P.; Pikus, E.; Whittingham, L.A.; Dunn, P.O. A global analysis of selection at the avian MHC. Evolution 2018, 72, 1278–1293. [Google Scholar] [CrossRef]
- Sommer, S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2005, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Pavey, S.A.; Bernatchez, L.; Aubin-Horth, N.; Landry, C.R. What is needed for next-generation ecological and evolutionary genomics? Trends Ecol. Evol. 2012, 27, 673–678. [Google Scholar] [CrossRef]
- O’Connor, E.A.; Westerdahl, H.; Burri, R.; Edwards, S.V. Avian MHC Evolution in the Era of Genomics: Phase 1.0. Cells 2019, 8, 1152. [Google Scholar] [CrossRef] [Green Version]
- Burri, R.; Salamin, N.; Studer, R.A.; Roulin, A.; Fumagalli, L. Adaptive divergence of ancient gene duplicates in the avian MHC class II β. Mol. Biol. Evol. 2010, 27, 2360–2374. [Google Scholar] [CrossRef] [Green Version]
- Ekblom, R.; Sæther, S.A.; Jacobsson, P.; Fiske, P.; Sahlman, T.; Grahn, M.; Kålås, J.A.; Höglund, J. Spatial pattern of MHC class II variation in the great snipe (Gallinago media). Mol. Ecol. 2007, 16, 1439–1451. [Google Scholar] [CrossRef]
- Walsh, H.E.; Friesen, V.L. A Comparison of Intraspecific Patterns of DNA Sequence Variation in Mitochondrial DNA, α-Enolase, and MHC Class II B Loci in Auklets (Charadriiformes: Alcidae). J. Mol. Evol. 2003, 57, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Cruz-López, M.; Fernández, G.; Hipperson, H.; Palacios, E.; Cavitt, J.; Galindo-Espinosa, D.; Gómez Del Angel, S.; Pruner, R.; Gonzalez, O.; Burke, T.; et al. Allelic diversity and patterns of selection at the major histocompatibility complex class I and II loci in a threatened shorebird, the Snowy Plover (Charadrius nivosus). BMC Evol. Biol. 2020, 20, 114. [Google Scholar] [CrossRef] [PubMed]
- Leclaire, S.; Van Dongen, W.F.D.; Voccia, S.; Merkling, T.; Ducamp, C.; Hatch, S.A.; Blanchard, P.; Danchin, É.; Wagner, R.H. Preen secretions encode information on MHC similarity in certain sex-dyads in a monogamous seabird. Sci. Rep. 2014, 4, 6920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velarde, E. Breeding biology of Heermann’s Gull on Isla Rasa, Gulf of California, Mexico. Auk 1999, 116, 513–519. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.; Ma, L.; Zhang, B.; Meng, G.; Xia, C. A Newly Recognized Pairing Mechanism of the α- and β-Chains of the Chicken Peptide–MHC Class II Complex. J. Immunol. 2020, 204, 1630–1640. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.H.; Jardetzky, T.S.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 1993, 364, 33–39. [Google Scholar] [CrossRef]
- Xu, R.; Li, K.; Chen, G.; Xu, H.; Qiang, B.; Li, C.; Liu, B. Characterization of Genetic Polymorphism of Novel MHC B-LB II Alleles in Chinese Indigenous Chickens. J. Genet. Genom. 2007, 34, 109–118. [Google Scholar] [CrossRef]
- Bowen, T.; Velarde, E.; Anderson, D.W.; Marlett, S.A. Federico Craveri and changes in nesting seabirds on Isla Rasa, Gulf of California, since 1856. Southwest. Nat. 2015, 60, 1–5. [Google Scholar] [CrossRef]
- Ruiz, E.A.; Velarde, E.; Aguilar, A. Demographic history of Heermann’s Gull (Larus heermanni) from late Quaternary to present: Effects of past climate change in the Gulf of California. Auk 2017, 134, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Mancilla-Morales, M.D.; Velarde, E.; Aguilar, A.; Contreras-Rodríguez, A.; Ezcurra, E.; Rosas-Rodríguez, J.A.; Soñanez-Organis, J.G.; Ruiz, E.A. Strong Philopatry, Isolation by Distance, and Local Habitat Have Promoted Genetic Structure in Heermann’s Gull. Diversity 2022, 14, 108. [Google Scholar] [CrossRef]
- Kikkawa, E.F.; Tsuda, T.T.; Naruse, T.K.; Sumiyama, D.; Fukuda, M.; Kurita, M.; Murata, K.; Wilson, R.P.; LeMaho, Y.; Tsuda, M.; et al. Analysis of the sequence variations in the Mhc DRB1-like gene of the endangered Humboldt penguin (Spheniscus humboldti). Immunogenetics 2005, 57, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.T.; Tsuda, M.; Naruse, T.; Kawata, H.; Ando, A.; Shiina, T.; Inoko, H. Phylogenetic analysis of penguin (Spheniscidae) species based on sequence variation in MHC class II genes. Immunogenetics 2001, 53, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Lenz, T.L.; Becker, S. Simple approach to reduce PCR artefact formation leads to reliable genotyping of MHC and other highly polymorphic loci-Implications for evolutionary analysis. Gene 2008, 427, 117–123. [Google Scholar] [CrossRef]
- Gouy, M.; Tannier, E.; Comte, N.; Parsons, D.P. Seaview Version 5: A Multiplatform Software for Multiple Sequence Alignment, Molecular Phylogenetic Analyses, and Tree Reconciliation. In Multiple Sequence Alignment; Katoh, K., Ed.; Humana: New York, NY, USA, 2021; Volume 2231, pp. 241–260. [Google Scholar] [CrossRef]
- BLAST. Basic Local Alignment Search Tool. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 19 May 2020).
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Ryvar, R.; Gaskell, R.; Addie, D.; Willoughby, K.; Carter, S.; Thomson, W.; Ollier, W.; Radford, A. Sequence analysis of MHC DRB alleles in domestic cats from the United Kingdom. Immunogenetics 2002, 54, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Bontrop, R.E.; Dawkins, R.L.; Erlich, H.A.; Gyllensten, U.B.; Heise, E.R.; Jones, P.P.; Parham, P.; Wakeland, E.K.; Watkins, D.I. Nomenclature for the major histocompatibility complexes of different species: A proposal. Immunogenetics 1990, 31, 217–219. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bryant, D.; Moulton, V. Neighbor-Net: An Agglomerative Method for the Construction of Phylogenetic Networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Furlong, R.F.; Yang, Z. Diversifying and purifying selection in the peptide binding region of DRB in mammals. J. Mol. Evol. 2008, 66, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protein Variability Server. Available online: http://imed.med.ucm.es/PVS/ (accessed on 23 March 2021).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. Available online: https://swissmodel.expasy.org/ (accessed on 19 July 2021). [CrossRef] [PubMed] [Green Version]
- RCSB Protein Data Bank. Available online: http://www.rcsb.org/ (accessed on 19 July 2021).
- Hughes, A.L.; Yeager, M. Natural selection at major histocompatibility complex loci of vertebrates. Annu. Rev. Genet. 1998, 32, 415–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. Available online: http://datamonkey.org/ (accessed on 15 August 2021). [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian AppRoximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [Green Version]
- Wertheim, J.O.; Murrell, B.; Smith, M.D.; Pond, S.L.K.; Scheffler, K. RELAX: Detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 2015, 32, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Onsins, S.E.; Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 2002, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez-Soriano, A.; Ramos-Onsins, S.E.; Rozas, J.; Calafell, F.; Navarro, A. Statistical power analysis of neutrality tests under demographic expansions, contractions and bottlenecks with recombination. Genetics 2008, 179, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [CrossRef]
- Schneider, S.; Excoffier, L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: Application to human mitochondrial DNA. Genetics 1999, 152, 1079–1089. [Google Scholar] [CrossRef]
- Harpending, H.C. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum. Biol. 1994, 66, 591–600. [Google Scholar]
- Eimes, J.A.; Lee, S.I.; Townsend, A.K.; Jablonski, P.; Nishiumi, I.; Satta, Y. Early duplication of a single MHC IIB locus prior to the passerine radiations. PLoS ONE 2016, 1, e0163456. [Google Scholar] [CrossRef] [Green Version]
- Eimes, J.A.; Townsend, A.K.; Sepil, I.; Nishiumi, I.; Satta, Y. Patterns of evolution of MHC class II genes of crows (Corvus) suggest trans-species polymorphism. PeerJ 2015, 3, e853. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Hordijk, W.; Gascuel, O. Improving the efficiency of SPR moves in phylogenetic tree search methods based on maximum likelihood. Bioinformatics 2005, 21, 4338–4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximim-Likelihood Phylogenies Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anmarkrud, J.A.; Johnsen, A.; Bachmann, L.; Lifjeld, J.T. Ancestral polymorphism in exon 2 of bluethroat (Luscinia svecica) MHC class II B genes. J. Evol. Biol. 2010, 23, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.K.M. Bootstrap hypothesis tests for evolutionary trees and other dendrograms. Proc. Natl. Acad. Sci. USA 1994, 91, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Dearborn, D.C.; Gager, A.B.; McArthur, A.G.; Gilmour, M.E.; Mandzhukova, E.; Mauck, R.A. Gene duplication and divergence produce divergent MHC genotypes without disassortative mating. Mol. Ecol. 2016, 25, 4355–4367. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. Applying the Bootstrap in Phylogeny Reconstruction. Stat. Sci. 2003, 18, 256–267. [Google Scholar] [CrossRef]
- The IUCN Red List of Threatened Species (Version 2018-1). Available online: www.iucnredlist.org (accessed on 21 July 2021).
- Bollmer, J.L.; Dunn, P.O.; Whittingham, L.A.; Wimpee, C. Extensive MHC class II B gene duplication in a passerine, the common yellowthroat (Geothlypis trichas). J. Hered. 2010, 101, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Bollmer, J.L.; Hull, J.M.; Ernest, H.B.; Sarasola, J.H.; Parker, P.G. Reduced MHC and neutral variation in the Galapagos hawk, an island endemic. BMC Evol. Biol. 2011, 11, 143. [Google Scholar] [CrossRef] [Green Version]
- Apanius, V.; Penn, D.; Slev, P.R.; Ramelle Ruff, L.; Potts, W.K. The nature of selection on the major histocompatibility complex. Crit. Rev. Immunol. 2017, 37, 75–120. [Google Scholar] [CrossRef]
- Veit, R.R.; Velarde, E.; Horn, M.H.; Manne, L.L. Population Growth and Long-Distance Vagrancy Leads to Colonization of Europe by Elegant Terns Thalasseus elegans. Front. Ecol. Evol. 2021, 9, 824. [Google Scholar] [CrossRef]
- Dias, M.P.; Martin, R.; Pearmain, E.J.; Burfield, I.J.; Small, C.; Phillips, R.A.; Yates, O.; Lascelles, B.; Borboroglu, P.G.; Croxall, J.P. Threats to seabirds: A global assessment. Biol. Conserv. 2019, 237, 525–537. [Google Scholar] [CrossRef]
- Velarde, E.; Ezcurra, E. Are seabirds’ life history traits maladaptive under present oceanographic variability? The case of Heermann’s Gull (Larus heermanni). Condor 2018, 120, 388–401. [Google Scholar] [CrossRef]
- Piatt, J.F.; Parrish, J.K.; Renner, H.M.; Schoen, S.K.; Jones, T.T.; Arimitsu, M.L.; Kuletz, K.J.; Bodenstein, B.; García-Reyes, M.; Duerr, R.S.; et al. Extreme mortality and reproductive failure of common murres resulting from the northeast Pacific marine heatwave of 2014–2016. PLoS ONE 2020, 15, e0226087. [Google Scholar] [CrossRef] [Green Version]
- Humphries, G.R.W.; Velarde, E.; Anderson, D.W.; Haase, B.; Sydeman, W.J. Seabirds as early warning indicators of climate events in the Pacific. PICES Press 2015, 23, 18–20. Available online: http://hdl.handle.net/1834/36370 (accessed on 27 March 2022).
- Vinkler, M.; Adelman, J.S.; Ardia, D.R. Chapter 20—Evolutionary and ecological immunology. In Avian Immunology, 3rd ed.; Kaspers, B., Schat, K.A., Göbel, T.W., Vervelde, L.B.T.-A.I., Eds.; Academic Press: Boston, MA, USA, 2022; pp. 519–557. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Sequences | Codons | S | K | π | dnt | daa |
|---|---|---|---|---|---|---|
| Larus heermanni (n = 57) | All (66) | 55 | 10.422 | 0.053 | 0.05 ± 0.01 | 0.12 ± 0.02 |
| PBR (17) | 22 | 5.243 | 0.103 | 0.10 ± 0.03 | 0.22 ± 0.06 | |
| Non-PBR (49) | 33 | 5.179 | 0.035 | 0.04 ± 0.01 | 0.09 ± 0.02 | |
| DAB cluster (n = 23) | All (66) | 28 | 4.980 | 0.025 | 0.03 ± 0.01 | 0.06 ± 0.01 |
| PBR (17) | 12 | 2.142 | 0.042 | 0.04 ± 0.01 | 0.11 ± 0.04 | |
| Non-PBR (49) | 16 | 2.838 | 0.019 | 0.02 ± 0.01 | 0.05 ± 0.01 | |
| DBB cluster (n = 34) | All (66) | 39 | 3.242 | 0.017 | 0.02 ± 0.00 | 0.04 ± 0.01 |
| PBR (17) | 13 | 1.230 | 0.024 | 0.02 ± 0.01 | 0.07 ± 0.02 | |
| Non-PBR (49) | 26 | 2.012 | 0.014 | 0.01 ± 0.00 | 0.03 ± 0.01 |
| Species | Codons | dN | dS | dN-dS | ω | dN ≠ dS | dN > dS | dN < dS |
|---|---|---|---|---|---|---|---|---|
| Larus heermanni (n = 57) | All (66) | 0.07 ± 0.02 | 0.02 ± 0.01 | 0.05 ± 0.02 | 3.50 | 2.37 (0.019) | 2.42 (0.009) | −2.44 (1) |
| PBR (20) | 0.19 ± 0.05 | 0.04 ± 0.03 | 0.15 ± 0.06 | 4.75 | 2.71 (0.008) | 2.61 (0.005) | −2.82 (1) | |
| Non PBR | 0.03 ± 0.01 | 0.02 ± 0.01 | 0.01 ± 0.02 | 1.50 | 0.56 (0.574) | 0.51 (0.306) | −0.542 (1) | |
| Rissa tridactyla (n = 53) | All | 0.12 ± 0.02 | 0.07 ± 0.02 | 0.05 ± 0.03 | 1.71 | 1.62 (0.109) | 1.73 (0.043) | −1.59 (1) |
| PBR | 0.36 ± 0.06 | 0.03 ± 0.02 | 0.33 ± 0.06 | 12.00 | 5.67 (***) | 5.39 (***) | −5.63 (1) | |
| non PBR | 0.04 ± 0.01 | 0.09 ± 0.03 | −0.05 ± 0.03 | 0.44 | −1.67 (0.098) | − 1.65 (1) | 1.71 (0.045) | |
| Pagophila eburnean (n = 2) | All | 0.20 ± 0.04 | 0.10 ± 0.04 | 0.10 ± 0.07 | 2.00 | 1.52 (0.132) | 1.42 (0.079) | −1.45 (1) |
| PBR | 0.62 ± 0.14 | 0.10 ± 0.08 | 0.53 ± 0.18 | 6.20 | 2.94 (0.004) | 2.93 (0.002) | −2.94 (1) | |
| Non PBR | 0.06 ± 0.03 | 0.10 ± 0.05 | −0.04 ± 0.06 | 0.60 | −0.65 (0.517) | −0.61 (1) | 0.63 (0.265) | |
| Sterna hirundo (n = 50) | All | 0.11 ± 0.03 | 0.05 ± 0.01 | 0.06 ± 0.03 | 2.20 | 2.44 (0.016) | 2.36 (0.010) | −2.44 (1) |
| PBR | 0.39 ± 0.09 | 0.04 ± 0.02 | 0.35 ± 0.08 | 9.75 | 3.92 (***) | 4.23 (***) | −4.20 (1) | |
| non PBR | 0.02 ± 0.01 | 0.05 ± 0.01 | −0.03 ± 0.02 | 0.40 | −2.06 (0.041) | −2.06 (1) | 2.07 (0.020) | |
| Aethia pusilla (n = 2) | All | 0.15 ± 0.05 | 0.00 ± 0.00 | 0.15 ± 0.05 | - | 2.62 (0.010) | 2.74 (0.004) | −2.65 (1) |
| PBR | 0.32 ± 0.19 | 0.00 ± 0.00 | 0.32 ± 0.18 | - | 1.78 (0.077) | 1.73 (0.043) | −1.68 (1) | |
| Non PBR | 0.08 ± 0.04 | 0.00 ± 0.00 | 0.08 ± 0.03 | - | 2.13 (0.035) | 2.11 (0.019) | −2.01 (1) | |
| Aethia cristatella (n = 2) | All | 0.05 ± 0.03 | 0.02 ± 0.02 | 0.03 ± 0.04 | 2.50 | 0.73 (0.464) | 0.78 (0.220) | −0.74 (1) |
| PBR | 0.12 ± 0.10 | 0.00 ± 0.00 | 0.12 ± 0.10 | - | 1.24 (0.216) | 1.19 (0.118) | −1.20 (1) | |
| Non PBR | 0.04 ± 0.03 | 0.04 ± 0.04 | 0.00 ± 0.05 | 1.00 | −0.07 (0.946) | −0.07 (1) | 0.07 (0.474) | |
| Brachyramphus marmoratus (n = 20) | All | 0.09 ± 0.02 | 0.11 ± 0.03 | −0.01 ± 0.04 | 3.00 | −0.31 (0.759) | −0.32 (1) | 0.31 (0.379) |
| PBR | 0.29 ± 0.07 | 0.08 ± 0.05 | 0.21 ± 0.09 | 3.63 | 2.30 (0.023) | 2.25 (0.013) | −2.33 (1) | |
| non PBR | 0.03 ± 0.01 | 0.12 ± 0.04 | −0.09 ± 0.04 | 0.25 | −2.10 (0.038) | −2.12 (1) | 2.15 (0.017) | |
| Alca torda (n = 16) | All | 0.06 ± 0.02 | 0.03 ± 0.01 | 0.04 ± 0.02 | 2.00 | 2.52 (0.013) | 2.46 (0.008) | −2.49 (1) |
| PBR | 0.18 ± 0.05 | 0.04 ± 0.03 | 0.13 ± 0.03 | 4.50 | 4.00 (***) | 3.91 (***) | −3.89 (1) | |
| Non PBR | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.00 ± 0.02 | 1.00 | 0.10 (0.918) | 0.10 (0.460) | −0.10 (1.00) | |
| Fratercula arctica (n = 19) | All | 0.09 ± 0.02 | 0.04 ± 0.02 | 0.05 ± 0.02 | 2.25 | 2.11 (0.037) | 2.08 (0.020) | −2.14 (1) |
| PBR | 0.26 ± 0.07 | 0.04 ± 0.03 | 0.22 ± 0.06 | 6.50 | 3.37 (0.001) | 3.57 (***) | −3.35 (1) | |
| Non PBR | 0.03 ± 0.01 | 0.04 ± 0.02 | −0.01 ± 0.02 | 0.75 | −0.61 (0.544) | −0.64 (1) | 0.64 (0.263) | |
| Uria aalge (n = 13) | All | 0.10 ± 0.02 | 0.07 ± 0.02 | 0.03 ± 0.03 | 1.42 | 1.24 (0.218) | 1.23 (0.112) | −1.23 (1) |
| PBR | 0.30 ± 0.07 | 0.08 ± 0.05 | 0.22 ± 0.08 | 3.75 | 2.90 (0.004) | 2.92 (0.002) | −2.82 (1) | |
| Non PBR | 0.03 ± 0.01 | 0.06 ± 0.02 | −0.03 ± 0.02 | 0.50 | −1.35 (0.179) | −1.37 (1) | 1.37 (0.087) | |
| Gallinago media (n = 35) | All | 0.09 ± 0.02 | 0.05 ± 0.02 | 0.04 ± 0.03 | 1.80 | 1.13 (0.259) | 1.16 (0.123) | −1.20 (1) |
| PBR | 0.29 ± 0.07 | 0.06 ± 0.04 | 0.23 ± 0.07 | 4.83 | 3.26 (0.002) | 3.35 (0.001) | −3.27 (1) | |
| Non PBR | 0.01 ± 0.00 | 0.05 ± 0.03 | −0.04 ± 0.03 | 0.20 | −1.14 (0.255) | −1.18 (1) | 1.17 (0.122) | |
| Limosa limosa (n = 4) | All | 0.16 ± 0.03 | 0.12 ± 0.03 | 0.05 ± 0.03 | 1.33 | 1.30 (0.195) | 1.31 (0.096) | −1.32 (1) |
| PBR | 0.31 ± 0.10 | 0.10 ± 0.06 | 0.21 ± 0.08 | 3.10 | 2.59 (0.011) | 2.75 (0.004) | −2.75 (1) | |
| Non PBR | 0.11 ± 0.02 | 0.13 ± 0.04 | −0.02 ± 0.04 | 0.84 | −0.46 (0.643) | −0.45 (1.00) | 0.45 (0.325) | |
| Philomachus pugnax (n = 4) | All | 0.16 ± 0.03 | 0.13 ± 0.04 | 0.02 ± 0.05 | 1.23 | 0.52 (0.604) | 0.49 (0.311) | −0.49 (1) |
| PBR | 0.27 ± 0.10 | 0.08 ± 0.08 | 0.19 ± 0.10 | 3.38 | 1.88 (0.063) | 1.84 (0.035) | −1.75 (1) | |
| Non PBR | 0.11 ± 0.03 | 0.16 ± 0.06 | −0.04 ± 0.05 | 0.69 | −0.78 (0.440) | −0.77 (1) | 0.79 (0.217) | |
| Charadrius nivosus (n = 4) | All | 0.04 ± 0.02 | 0.07 ± 0.03 | −0.03 ± 0.03 | 0.57 | −1.11 (0.271) | −1.12 (1) | 1.35 (0.129) |
| PBR | 0.10 ± 0.05 | 0.06 ± 0.05 | 0.04 ± 0.03 | 1.66 | 1.26 (0.211) | 1.30 (0.099) | −1.26 (1) | |
| Non PBR | 0.02 ± 0.01 | 0.08 ± 0.04 | −0.06 ± 0.04 | 0.25 | −1.54 (0.124) | −1.66 (1) | 1.71 (0.045) |
| Reference Branches | k | Selection | p | IUCN |
|---|---|---|---|---|
| Rissa tridactyla (71) | 0 | relaxed | 0.000 | Vulnerable |
| Sterna hirundo (55) | 0 | relaxed | 0.000 | Least concern |
| Pagophila eburnea (5) | 1.11 | intensified | 0.773 | Near threatened |
| Aethia pusilla (2) | 0.28 | relaxed | 0.016 | Least concern |
| Aethia cristatella (2) | 1.01 | intensified | 0.979 | Least concern |
| Brachyramphus marmoratus (27) | 1.77 | intensified | 0.126 | Endangered |
| Alca torda (24) | 0.78 | relaxed | 0.342 | Near threatened |
| Fratercula arctica (33) | 0.71 | relaxed | 0.048 | Vulnerable |
| Uria aalge (14) | 0.55 | relaxed | 0.093 | Least concern |
| Gallinago media (37) | 0.44 | relaxed | 0.000 | Near threatened |
| Limosa limosa (4) | 1.14 | intensified | 0.617 | Near threatened |
| Philomachus pugnax (6) | 0.75 | relaxed | 0.201 | Least concern |
| Charadrius niveus (6) | 0.32 | relaxed | 0.000 | Near threatened |
| Genes | π | Hd | D | Fs | R2 | τ | t | rg | SSD |
|---|---|---|---|---|---|---|---|---|---|
| MHCIIB | 0.0473 | 0.82 | −0.16 ns | −21.70 * | 0.08 ns | 19.82 (6.44–108.83) | 28 to 31 yr BP (9–172 yr BP) | 0.04397 p = 0.1564 | 0.06017 p = 0.0396 |
| Cyt-b | 0.0009 | 0.63 | −2.12 * | −11.06 * | 0.04 * | 1 (0.74–1.86) | 48,403 to 53,781 yr BP (35,924–100,211 yr BP) | 0.06358 p = 0.1782 | 0.00086 p = 0.6119 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancilla-Morales, M.D.; Velarde, E.; Contreras-Rodríguez, A.; Gómez-Lunar, Z.; Rosas-Rodríguez, J.A.; Heras, J.; Soñanez-Organis, J.G.; Ruiz, E.A. Characterization, Selection, and Trans-Species Polymorphism in the MHC Class II of Heermann’s Gull (Charadriiformes). Genes 2022, 13, 917. https://doi.org/10.3390/genes13050917
Mancilla-Morales MD, Velarde E, Contreras-Rodríguez A, Gómez-Lunar Z, Rosas-Rodríguez JA, Heras J, Soñanez-Organis JG, Ruiz EA. Characterization, Selection, and Trans-Species Polymorphism in the MHC Class II of Heermann’s Gull (Charadriiformes). Genes. 2022; 13(5):917. https://doi.org/10.3390/genes13050917
Chicago/Turabian StyleMancilla-Morales, Misael Daniel, Enriqueta Velarde, Araceli Contreras-Rodríguez, Zulema Gómez-Lunar, Jesús A. Rosas-Rodríguez, Joseph Heras, José G. Soñanez-Organis, and Enrico A. Ruiz. 2022. "Characterization, Selection, and Trans-Species Polymorphism in the MHC Class II of Heermann’s Gull (Charadriiformes)" Genes 13, no. 5: 917. https://doi.org/10.3390/genes13050917
APA StyleMancilla-Morales, M. D., Velarde, E., Contreras-Rodríguez, A., Gómez-Lunar, Z., Rosas-Rodríguez, J. A., Heras, J., Soñanez-Organis, J. G., & Ruiz, E. A. (2022). Characterization, Selection, and Trans-Species Polymorphism in the MHC Class II of Heermann’s Gull (Charadriiformes). Genes, 13(5), 917. https://doi.org/10.3390/genes13050917