
Application of Dual Metabarcoding Platforms for the Meso- and Macrozooplankton Taxa in the Ross Sea

 , ,
, ,  ,
,  , and
, and 
Abstract
:1. Introduction
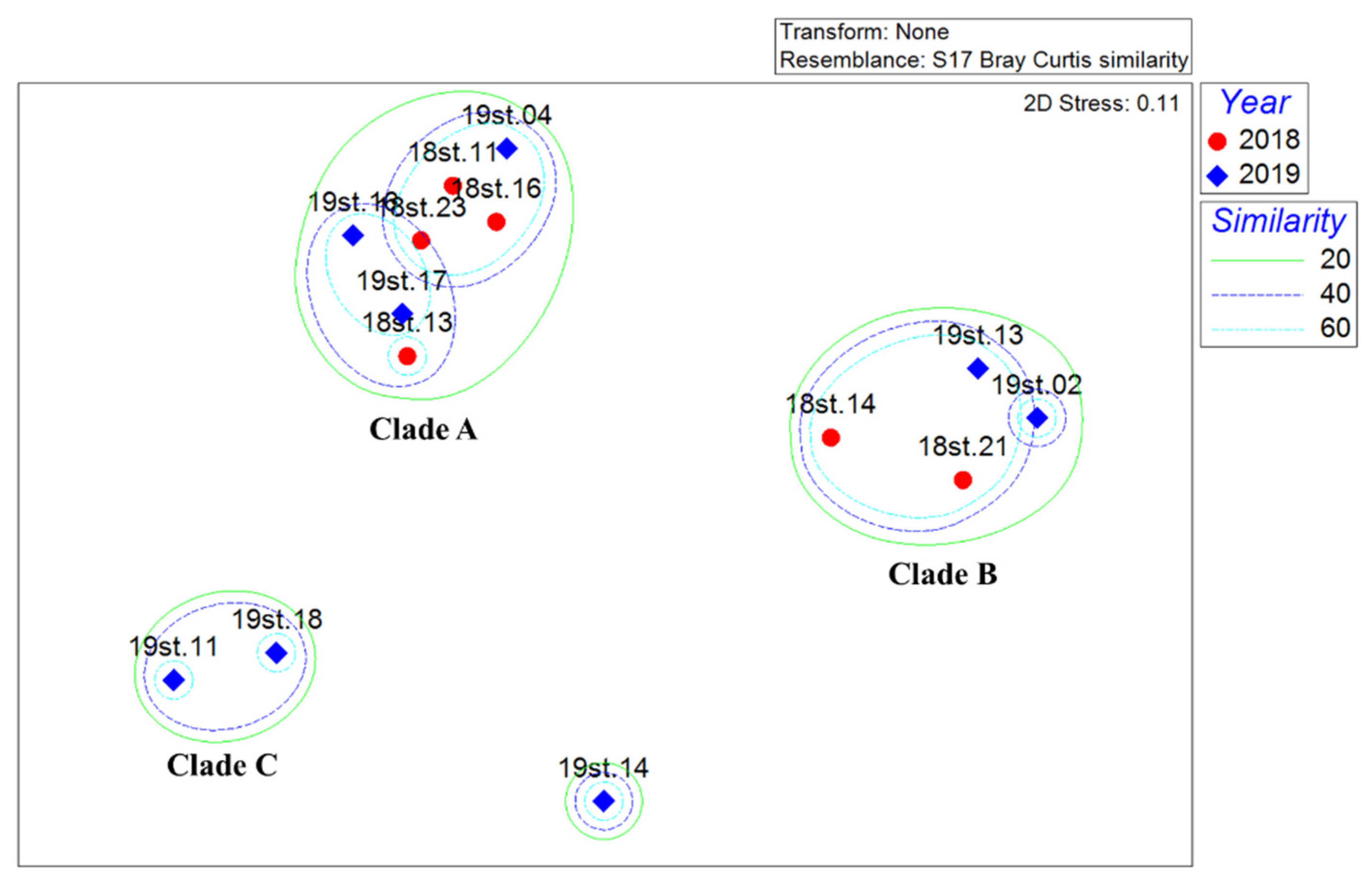
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. PacBio Amplicon Sequencing for Regular Barcodes (RBs)
2.3. Illumina Miseq Sequencing for Mini Barcodes (MBs)
2.4. Bioinformatic Analysis of Regular Barcodes (RBs)
2.5. Bioinformatic Analysis of Mini Barcodes (MBs)
3. Results
3.1. Extraction of Haplotypes from Regular Barcodes (RBs) and Mini Barcodes (MBs)
3.2. Annelida
3.3. Arthropoda
3.4. Mollusca
3.5. Nemertea
3.6. Others
3.7. Spatiotemporal Distribution of Zooplankton Species Using Metabarcoding Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaufman, D.E.; Friedrichs, M.A.M.; Smith, W.O., Jr.; Hofmann, E.E.; Dinniman, M.S.; Hemmings, J.C.P. Climate change impacts on southern Ross Sea phytoplankton composition, productivity, and export. J. Geophys. Res. 2017, 122, 2339–2359. [Google Scholar] [CrossRef]
- Convey, P.; Peck, L.S. Antarctic environmental change and biological responses. Sci. Adv. 2019, 5, eaaz0888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hays, G.C.; Richardson, A.J.; Robinson, C. Climate change and marine plankton. Trends Ecol. Evol. 2005, 20, 337–344. [Google Scholar] [CrossRef] [PubMed]
- McBride, M.M.; Dalpadado, P.; Drinkwater, K.F.; Godø, O.R.; Hobday, A.J.; Hollowed, A.B.; Kristiansen, T.; Murphy, E.J.; Ressler, P.H.; Subbey, S. Krill, climate, and contrasting future scenarios for Arctic and Antarctic fisheries. ICES Mar. Sci. Symp. 2014, 71, 1934–1955. [Google Scholar] [CrossRef] [Green Version]
- Constable, A.J.; Melbourne-Thomas, J.; Corney, S.P.; Arrigo, K.R.; Barbraud, C.; Barnes, D.K.; Bindoff, N.L.; Boyd, P.W.; Brandt, A.; Costa, D.P. Climate change and Southern Ocean ecosystems I: How changes in physical habitats directly affect marine biota. Glob. Chang. Biol. 2014, 20, 3004–3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Li, C.; Sun, S. Inter-annual variation in summer zooplankton community structure in Prydz Bay, Antarctica, from 1999 to 2006. Polar Biol. 2011, 34, 921–932. [Google Scholar] [CrossRef]
- Moriarty, R.; Buitenhuis, E.; Le Quéré, C.; Gosselin, M.-P. Distribution of known macrozooplankton abundance and biomass in the global ocean. Earth Syst. Sci. Data 2013, 5, 241–257. [Google Scholar] [CrossRef] [Green Version]
- Tagliabue, A.; Arrigo, K.R. Anomalously low zooplankton abundance in the Ross Sea: An alternative explanation. Limnol. Oceanogr. 2003, 48, 686–699. [Google Scholar] [CrossRef]
- Steinberg, D.K.; Ruck, K.E.; Gleiber, M.R.; Garzio, L.M.; Cope, J.S.; Bernard, K.S.; Stammerjohn, S.E.; Schofield, O.M.; Quetin, L.B.; Ross, R.M. Long-term (1993–2013) changes in macrozooplankton off the Western Antarctic Peninsula. Deep. Sea Res. Part I Oceanogr. Res. Pap. 2015, 101, 54–70. [Google Scholar] [CrossRef]
- Criales-Hernández, M.I.; Jerez-Guerrero, M.; Latandret-Solana, S.A.; Gómez-Sánchez, M.D. Spatial distribution of meso-and macro-zooplankton in the Bransfield Strait and around Elephant Island, Antarctic Peninsula, during the 2019–2020 austral summer. Polar Sci. 2022, 100821. [Google Scholar] [CrossRef]
- Dietrich, K.S.; Santora, J.A.; Reiss, C.S. Winter and summer biogeography of macrozooplankton community structure in the northern Antarctic Peninsula ecosystem. Prog. Oceanogr. 2021, 196, 102610. [Google Scholar] [CrossRef]
- Hosie, G.; Fukuchi, M.; Kawaguchi, S. Development of the Southern Ocean continuous plankton recorder survey. Prog. Oceanogr. 2003, 58, 263–283. [Google Scholar] [CrossRef]
- Pinkerton, M.H.; Décima, M.; Kitchener, J.A.; Takahashi, K.T.; Robinson, K.V.; Stewart, R.; Hosie, G.W. Zooplankton in the Southern Ocean from the continuous plankton recorder: Distributions and long-term change. Deep-Sea Res. I Oceanogr. Res. Pap. 2020, 162, 103303. [Google Scholar] [CrossRef]
- Bucklin, A.; Lindeque, P.K.; Rodriguez-Ezpeleta, N.; Albaina, A.; Lehtiniemi, M. Metabarcoding of marine zooplankton: Prospects, progress and pitfalls. J. Plankton Res. 2016, 38, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Morales, E.A.; Trainor, F.R. Algal Phenotypic Plasticity: Its Importance in Developing New Concepts The Case for Scenedesmus. ALGAE 1997, 12, 147–157. [Google Scholar]
- Yang, J.; Zhang, X. eDNA metabarcoding in zooplankton improves the ecological status assessment of aquatic ecosystems. Environ. Int. 2020, 134, 105230. [Google Scholar] [CrossRef]
- Dunthorn, M.; Otto, J.; Berger, S.A.; Stamatakis, A.; Mahé, F.; Romac, S.; de Vargas, C.; Audic, S.; Consortium, B.; Stock, A.; et al. Placing Environmental Next-Generation Sequencing Amplicons from Microbial Eukaryotes into a Phylogenetic Context. Mol. Biol. Evol. 2014, 31, 993–1009. [Google Scholar] [CrossRef] [Green Version]
- Mahé, F.; Mayor, J.; Bunge, J.; Chi, J.; Siemensmeyer, T.; Stoeck, T.; Wahl, B.; Paprotka, T.; Filker, S.; Dunthorn, M. Comparing High-throughput Platforms for Sequencing the V4 Region of SSU-rDNA in Environmental Microbial Eukaryotic Diversity Surveys. J. Eukaryot. Microbiol. 2015, 62, 338–345. [Google Scholar] [CrossRef]
- Martijn, J.; Lind, A.E.; Schön, M.E.; Spiertz, I.; Juzokaite, L.; Bunikis, I.; Pettersson, O.V.; Ettema, T.J.G. Confident phylogenetic identification of uncultured prokaryotes through long read amplicon sequencing of the 16S-ITS-23S rRNA operon. Environ. Microbiol. 2019, 21, 2485–2498. [Google Scholar] [CrossRef]
- Heeger, F.; Bourne, E.C.; Baschien, C.; Yurkov, A.; Bunk, B.; Spröer, C.; Overmann, J.; Mazzoni, C.J.; Monaghan, M.T. Long-read DNA metabarcoding of ribosomal RNA in the analysis of fungi from aquatic environments. Mol. Ecol. Resour. 2018, 18, 1500–1514. [Google Scholar] [CrossRef]
- Heimeier, D.; Lavery, S.; Sewell, M.A. Using DNA barcoding and phylogenetics to identify Antarctic invertebrate larvae: Lessons from a large scale study. Mar. Genom. 2010, 3, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Gallego, R.; Heimeier, D.; Lavery, S.; Sewell, M.A. The meroplankton communities from the coastal Ross Sea: A latitudinal study. Hydrobiologia 2015, 761, 195–209. [Google Scholar] [CrossRef]
- Smith, W.O., Jr.; Ainley, D.G.; Arrigo, K.R.; Dinniman, M.S. The Oceanography and Ecology of the Ross Sea. Ann. Rev. Mar. Sci. 2014, 6, 469–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, G.; Jongsomjit, D.; Veloz, S.; Ainley, D. Coexistence of mesopredators in an intact polar ocean ecosystem: The basis for defining a Ross Sea Marine Protected Area. Biol. Conserv. 2011, 156. [Google Scholar] [CrossRef]
- McEwen, G.F.; Johnson, M.; Folsom, T.R. A statistical analysis of the performance of the Folsom plankton sample splitter, based upon test observations. Arch. Für. Meteorol. Geophys. Bioklimatol. Ser. A 1954, 7, 502–527. [Google Scholar] [CrossRef]
- Geller, J.; Meyer, C.; Parker, M.; Hawk, H. Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol. Ecol. Resour. 2013, 13, 851–861. [Google Scholar] [CrossRef]
- Yoon, T.-H.; Kang, H.-E.; Lee, S.R.; Lee, J.-B.; Baeck, G.W.; Park, H.; Kim, H.-W. Metabarcoding analysis of the stomach contents of the Antarctic Toothfish (Dissostichus mawsoni) collected in the Antarctic Ocean. PeerJ 2017, 5, e3977. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Version 4.0. 0; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Elbrecht, V.; Vamos, E.E.; Meissner, K.; Aroviita, J.; Leese, F. Assessing strengths and weaknesses of DNA metabarcoding-based macroinvertebrate identification for routine stream monitoring. Methods Ecol. Evol. 2017, 8, 1265–1275. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Frøslev, T.G.; Kjøller, R.; Bruun, H.H.; Ejrnæs, R.; Brunbjerg, A.K.; Pietroni, C.; Hansen, A.J. Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.; Gorley, R. Getting started with PRIMER v7. PRIMER-E Plymouth Plymouth Mar. Lab. 2015, 20, 1–20. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Kiko, R.; Kramer, M.; Spindler, M.; Wägele, H. Tergipes antarcticus (Gastropoda, Nudibranchia): Distribution, life cycle, morphology, anatomy and adaptation of the first mollusc known to live in Antarctic sea ice. Polar Biol. 2008, 31, 1383–1395. [Google Scholar] [CrossRef]
- Riccardi, N. Selectivity of plankton nets over mesozooplankton taxa: Implications for abundance, biomass and diversity estimation. J. Limnol. 2010, 69, 287. [Google Scholar] [CrossRef]
- Gallienne, C.P.; Robins, D.B. Is Oithona the most important copepod in the world’s oceans? J. Plankton Res. 2001, 23, 1421–1432. [Google Scholar] [CrossRef]
- Greene, C.H. A brief review and critique of zooplankton sampling methods: Copepodology for the larval ecologist. Ophelia 1990, 32, 109–113. [Google Scholar] [CrossRef]
- Makabe, R.; Tanimura, A.; Fukuchi, M. Comparison of mesh size effects on mesozooplankton collection efficiency in the Southern Ocean. J. Plankton Res. 2012, 34, 432–436. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, A. Life cycle strategies of epipelagic copepods in the Southern Ocean. J. Mar. Syst. 1998, 15, 289–311. [Google Scholar] [CrossRef]
- Eastman, J.T. The nature of the diversity of Antarctic fishes. Polar Biol. 2005, 28, 93–107. [Google Scholar] [CrossRef]
- North, A.W.; Kellermann, A. Key to the early stages of Antarctic fish. Ber Polarforsch 1990, 67, 1–44. [Google Scholar]
- Eastman, J.T.; Devries, A.L. Biology and phenotypic plasticity of the Antarctic nototheniid fish Trematomus newnesi in McMurdo Sound. Antarct. Sci. 2004, 9, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Matschiner, M.; Hanel, R.; Salzburger, W. On the origin and trigger of the notothenioid adaptive radiation. PLoS ONE 2011, 6, e18911. [Google Scholar] [CrossRef]
- Near, T.J. Notothenioid fishes (Notothenioidei). In The Timetree of Life; Oxford University Press: New York, NY, USA, 2009; pp. 339–343. [Google Scholar]
- Antich, A.; Palacin, C.; Wangensteen, O.S.; Turon, X. To denoise or to cluster, that is not the question: Optimizing pipelines for COI metabarcoding and metaphylogeography. BMC Bioinform. 2021, 22, 177. [Google Scholar] [CrossRef]
- Bonello, G.; Grillo, M.; Cecchetto, M.; Giallain, M.; Granata, A.; Guglielmo, L.; Pane, L.; Schiaparelli, S. Distributional records of Ross Sea (Antarctica) planktic Copepoda from bibliographic data and samples curated at the Italian National Antarctic Museum (MNA): Checklist of species collected in the Ross Sea sector from 1987 to 1995. ZooKeys 2020, 969, 1–22. [Google Scholar] [CrossRef]
- Smith, W.O.; Delizo, L.M.; Herbolsheimer, C.; Spencer, E. Distribution and abundance of mesozooplankton in the Ross Sea, Antarctica. Polar Biol. 2017, 40, 2351–2361. [Google Scholar] [CrossRef]
- Stevens, C.J.; Pakhomov, E.A.; Robinson, K.V.; Hall, J.A. Mesozooplankton biomass, abundance and community composition in the Ross Sea and the Pacific sector of the Southern Ocean. Polar Biol. 2015, 38, 275–286. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regular Barcoding (RB) | Minibarcoding (MB) | |||||
|---|---|---|---|---|---|---|
| Denoised ASVs (Abundant) | Clustered OTUs (Rare) | Denoised ASVs | ||||
| 2018 | 2019 | 2018 | 2019 | 2018 | 2019 | |
| PacBio CCS reads | 42,552 | 34,075 | 42,552 | 34,075 | n/a | n/a |
| MiSeq reads | n/a | n/a | n/a | n/a | 2,640,586 | 788,096 |
| Denoised reads | 14,341 (33.7%) | 11,923 (35.0%) | n/a | n/a | 1,268,522 (48.0%) | 360,336 (45.7%) |
| Clustered reads | n/a | n/a | 41,119 (96.6%) | 33,013 (96.9%) | n/a | n/a |
| Amplicon sequence variants (ASVs) | 26 | 45 | n/a | n/a | 206 | 122 |
| Operational taxonomic units (OTUs) | n/a | n/a | 37,778 | 26,069 | n/a | n/a |
| Metazoan ASVs (reads) | 18 (14,278) | 32 (10,798) | 154 (1,151,931) | 103 (232,332) | ||
| Non-metazoan ASVs (reads) | 8 (63) | 13 (1125) | 52 (116,591) | 19 (128,004) | ||
| Putative haplotypes (reads) | 46 ASVs (25,076) | 9 OTUs (240) | 183 ASVs (1,384,263) | |||
| Number of phyla | 5 | 8 | ||||
| Number of genera | 20 | 32 | ||||
| Phylum | Class | Order | Family | Description | February 2018 | January 2019 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| st. 11 | st. 13 | st. 14 | st. 16 | st. 21 | st. 23 | Avg. | st. 2 | st. 4 | st. 11 | st. 13 | st. 14 | st. 16 | st. 17 | st. 18 | Avg. | |||||
| Annelida | Polychaeta | Amphinomida | Amphinomidae | Amphinomidae sp. | 0.22 | 2.58 | 0.03 | 2.28 | 0.00 | 0.85 | 0.00 | 0.00 | 0.00 | |||||||
| Phyllodocida | Phyllodocidae | Phyllodocidae sp. | 1.31 | 0.22 | 0.00 | |||||||||||||||
| Polynoidae | Polynoidae sp. | 0.01 | 0.00 | 0.00 | ||||||||||||||||
| Spionida | Spionidae | L. antarctica | 0.00 | 0.04 | 0.01 | 0.00 | ||||||||||||||
| Laonice sp. | 0.02 | 0.00 | 0.00 | |||||||||||||||||
| Laonice weddellia | 0.05 | 0.01 | 0.03 | 0.00 | 0.00 | 0.01 | 0.00 | |||||||||||||
| S. eltaninae | 0.42 | 0.33 | 0.56 | 10.30 | 1.93 | 4.59 | 0.01 | 5.34 | 0.02 | 0.01 | 1.25 | |||||||||
| Spiophanes sp. | 0.01 | 0.05 | 0.01 | 0.00 | ||||||||||||||||
| Arthropoda | Hexanauplia | Calanoida | Calanidae | C. acutus | 3.02 | 3.35 | 2.93 | 3.36 | 1.73 | 1.56 | 2.66 | 1.79 | 0.02 | 0.01 | 96.37 | 1.11 | 16.78 | 5.11 | 15.15 | |
| Calanus propinquus | 1.02 | 0.33 | 0.01 | 0.26 | 0.02 | 0.58 | 0.37 | 0.38 | 0.01 | 0.60 | 1.69 | 9.43 | 1.51 | |||||||
| Calanus simillimus | 0.05 | 0.02 | 0.00 | 0.00 | 0.00 | 0.01 | 0.11 | 0.52 | 0.08 | |||||||||||
| Calanidae sp. | 0.01 | 0.00 | 0.00 | |||||||||||||||||
| Clausocalanidae | C. citer | 14.55 | 6.24 | 6.52 | 0.00 | 0.01 | 0.33 | 4.61 | 0.51 | 0.06 | ||||||||||
| Euchaetidae | P. antarctica | 0.13 | 22.60 | 0.18 | 0.75 | 0.01 | 1.32 | 4.16 | 0.26 | 0.00 | 0.25 | 0.19 | 0.01 | 0.09 | ||||||
| Metridinidae | M. gerlachei | 64.51 | 7.40 | 4.20 | 53.03 | 0.03 | 54.01 | 30.53 | 70.28 | 0.03 | 0.01 | 2.09 | 32.38 | 19.12 | 8.13 | 16.50 | ||||
| Rhincalanidae | Rhincalanus gigas | 0.09 | 0.02 | 0.12 | 0.01 | |||||||||||||||
| Scolecitrichidae | Scolecitrichidae sp. | 0.01 | 0.00 | 0.00 | ||||||||||||||||
| Cyclopoida | Oithonidae | Oithona frigida | 0.01 | 0.06 | 0.40 | 0.01 | 0.16 | 0.11 | 0.02 | 0.00 | ||||||||||
| O. similis | 0.17 | 0.06 | 0.01 | 0.04 | 0.00 | |||||||||||||||
| Thecostraca | Balanomorpha | Bathylasmatidae | Bathylasma corolliforme | 0.00 | 0.00 | 0.00 | ||||||||||||||
| Malacostraca | Amphipoda | Hyperiidae | Hyperiella dilatata | 8.20 | 1.37 | 0.00 | ||||||||||||||
| Tryphosidae | Pseudorchomene plebs | 0.12 | 0.02 | 1.79 | 50.31 | 0.00 | 0.01 | 6.51 | ||||||||||||
| Pseudorchomene sp. | 0.00 | 0.02 | 0.00 | |||||||||||||||||
| Euphausiacea | Euphausiidae | Euphausia crystallorophias | 0.00 | 0.01 | 0.02 | 1.08 | 0.19 | 61.27 | 1.02 | 0.01 | 20.90 | 0.03 | 0.00 | 10.41 | ||||||
| E. superba | 0.47 | 0.09 | 1.10 | 0.13 | 6.97 | 1.46 | 0.08 | 2.68 | 0.04 | 0.25 | 0.01 | 0.28 | 0.55 | 3.23 | 0.89 | |||||
| Thysanoessa macrura | 1.09 | 0.18 | 0.00 | |||||||||||||||||
| Ostracoda | Halocyprida | Halocyprididae | Alacia hettacra | 2.73 | 6.12 | 0.15 | 9.19 | 0.00 | 12.84 | 5.17 | 2.42 | 0.11 | 0.14 | 0.33 | ||||||
| Austrinoecia isocheira | 0.09 | 0.04 | 0.00 | 0.06 | 1.68 | 0.31 | 0.01 | 0.00 | ||||||||||||
| Boroecia antipoda | 0.02 | 0.00 | 0.00 | |||||||||||||||||
| Chaetognatha | Sagittoidea | Phragmophora | Eukrohniidae | Eukrohniidae sp. | 0.00 | 0.00 | 0.00 | |||||||||||||
| Chordata | Actinopterygii | Perciformes | Nototheniidae | P. antarctica | 0.00 | 5.95 | 0.74 | |||||||||||||
| Cnidaria | Hydrozoa | Hydrozoa sp. | 3.46 | 0.15 | 0.44 | 0.67 | 0.00 | |||||||||||||
| Siphonophorae | Sphaeronectidae | Sphaeronectidae sp. | 0.02 | 0.01 | 0.00 | 0.23 | 0.04 | 0.00 | ||||||||||||
| Echinodermata | Asteroidea | Valvatida | Odontasteridae | Odontaster meridionalis | 0.04 | 0.01 | 0.00 | |||||||||||||
| Mollusca | Gastropoda | Neogastropoda | Conidae | Conus sp. | 0.02 | 0.00 | 0.00 | |||||||||||||
| Nudibranchia | Tergipedidae | T. antarcticus | 14.71 | 2.45 | 0.00 | |||||||||||||||
| Pteropoda | Cliidae | Clio pyramidata | 0.13 | 0.02 | 0.64 | 42.59 | 0.00 | 0.00 | 0.04 | 0.05 | 73.58 | 14.61 | ||||||||
| Clionidae | C. limacina antarctica | 0.01 | 3.26 | 80.75 | 0.04 | 96.87 | 0.01 | 30.16 | 32.19 | 0.26 | 0.07 | 68.67 | 0.02 | 0.04 | 0.00 | 12.65 | ||||
| Limacinidae | Limacina rangii | 0.08 | 0.22 | 0.27 | 0.10 | 0.00 | ||||||||||||||
| Pneumodermatidae | Spongiobranchaea sp. | 0.20 | 0.03 | 0.03 | 17.33 | 2.17 | ||||||||||||||
| Nemertea | Pilidiophora | Heteronemertea | Lineidae | Lineus sp. | 0.02 | 0.00 | 0.00 | |||||||||||||
| Parvicirrus sp. | 0.82 | 0.03 | 0.01 | 4.52 | 0.09 | 0.34 | 0.97 | 0.51 | 13.27 | 0.12 | 4.81 | 0.37 | 0.13 | 2.40 | ||||||
| Unknown | Unknown | Unknown | Unknown | Unknown_Annelida | 0.00 | 0.00 | 0.00 | |||||||||||||
| Unknown_Arthropoda | 0.04 | 0.01 | 0.01 | 0.01 | 0.03 | 0.00 | ||||||||||||||
| Unknown_Bryozoa | 0.00 | 0.00 | 0.00 | |||||||||||||||||
| Unknown_Cnidaria | 0.00 | 45.34 | 2.72 | 0.29 | 0.04 | 19.01 | 11.23 | 0.13 | 6.36 | 0.00 | 0.90 | 48.06 | 60.01 | 0.00 | 14.43 | |||||
| Unknown_Mollusca | 0.01 | 0.00 | 0.26 | 0.05 | 0.00 | |||||||||||||||
| Unknown_Nematoda | 0.00 | 0.24 | 0.03 | |||||||||||||||||
| Unknown_Nemertea | 0.01 | 0.00 | 1.15 | 0.14 | ||||||||||||||||
| Unknown_Porifera | 0.01 | 0.00 | 0.00 | |||||||||||||||||
| Number of Genus | 18 | 21 | 16 | 16 | 14 | 15 | 16.7 | 4 | 12 | 13 | 9 | 7 | 8 | 14 | 6 | 9.1 | ||||
| Number of Species | 24 | 26 | 19 | 18 | 15 | 17 | 19.8 | 5 | 13 | 14 | 10 | 7 | 8 | 16 | 8 | 10.1 | ||||
| Platform | Shared Genera or Families | Regular-Read Bacodes (RB) | Mini-Read Barcodes (MB) | |
|---|---|---|---|---|
| Phylum | ||||
| Annelida | Amphinomidae Vanadis | Polynoidae (0%) Laonice (0%) Spiophanes (0.01%) | ||
| Arthropoda | * Calanoides Ctenocalanus * Paraeuchaeta Metridia Bathylasma * Oithona Pseudorchomene Euphausia | * Nematocarcinus (0%) | Calanus (1.07%) Rhincalanus (0.02%) Scolecitrichidae (0%) Hyperiella (0.59%) Thysanoessa (0.08%) Alacia (2.41%) Austrinoecia (0.13%) Boroecia (0%) | |
| Chaetognatha | Eukrohniidae (0%) | |||
| Chordata | Pleuragramma | Notolepis (0.31%) | ||
| Cnidaria | Sphaeronectes (0.02%) | |||
| Echinodermata | Odontaster (0%) | |||
| Mollusca | * Tergipes Clio Clione Pneumodermatidae | Cryocapulus (0.22%) | Conus (0%) Limacina (0.04%) | |
| Nemertea | Parvicirrus | Lineus (0%) | ||
| Total genera | 17 | 3 | 17 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-H.; La, H.S.; Kim, J.-H.; Son, W.; Park, H.; Kim, Y.-M.; Kim, H.-W. Application of Dual Metabarcoding Platforms for the Meso- and Macrozooplankton Taxa in the Ross Sea. Genes 2022, 13, 922. https://doi.org/10.3390/genes13050922
Lee J-H, La HS, Kim J-H, Son W, Park H, Kim Y-M, Kim H-W. Application of Dual Metabarcoding Platforms for the Meso- and Macrozooplankton Taxa in the Ross Sea. Genes. 2022; 13(5):922. https://doi.org/10.3390/genes13050922
Chicago/Turabian StyleLee, Ji-Hyun, Hyoung Sul La, Jeong-Hoon Kim, Wuju Son, Hyun Park, Young-Mog Kim, and Hyun-Woo Kim. 2022. "Application of Dual Metabarcoding Platforms for the Meso- and Macrozooplankton Taxa in the Ross Sea" Genes 13, no. 5: 922. https://doi.org/10.3390/genes13050922
APA StyleLee, J. -H., La, H. S., Kim, J. -H., Son, W., Park, H., Kim, Y. -M., & Kim, H. -W. (2022). Application of Dual Metabarcoding Platforms for the Meso- and Macrozooplankton Taxa in the Ross Sea. Genes, 13(5), 922. https://doi.org/10.3390/genes13050922