
Tissue-Specific Variations in Transcription Factors Elucidate Complex Immune System Regulation


Abstract
:1. Introduction
2. Materials and Methods
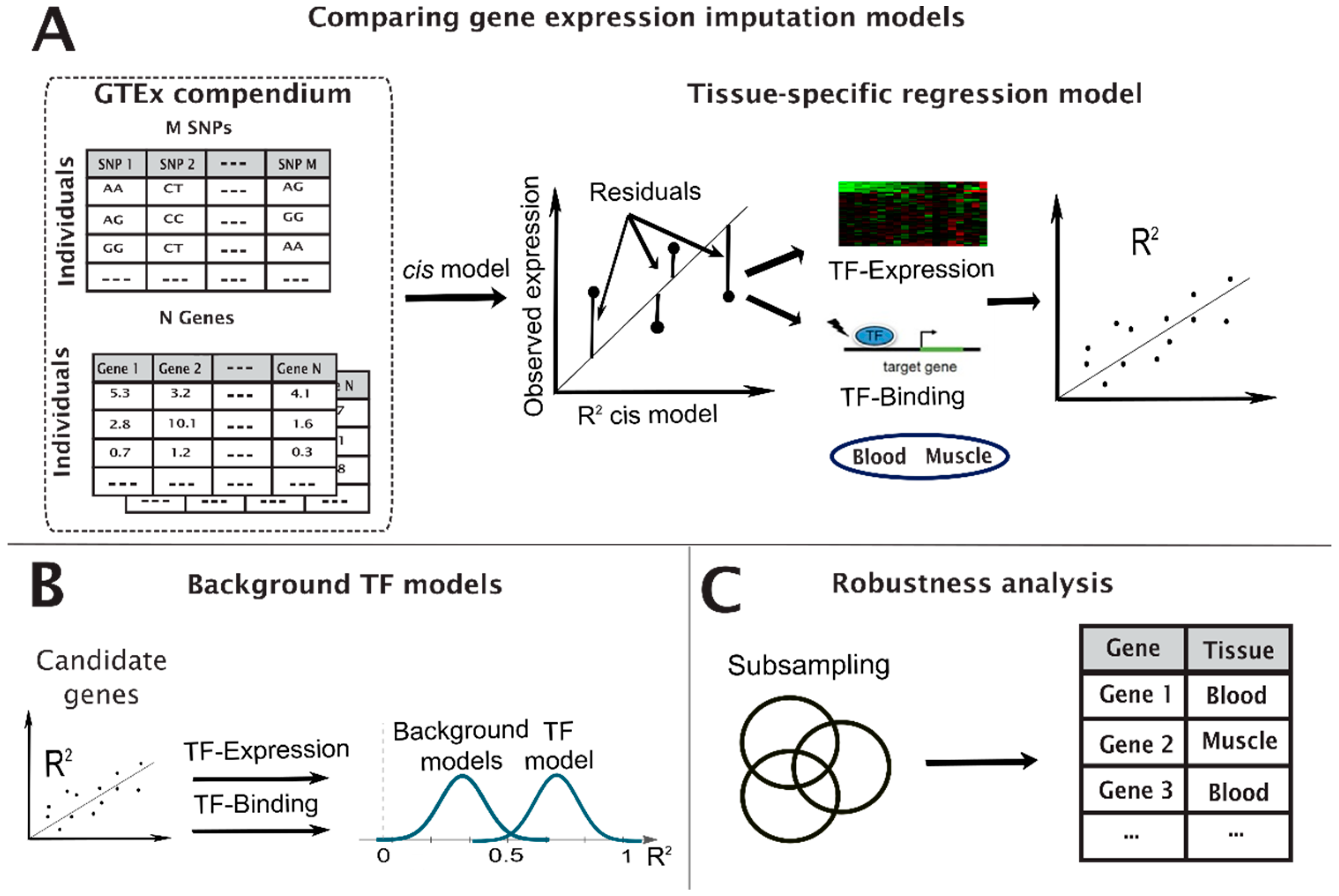
2.1. Data
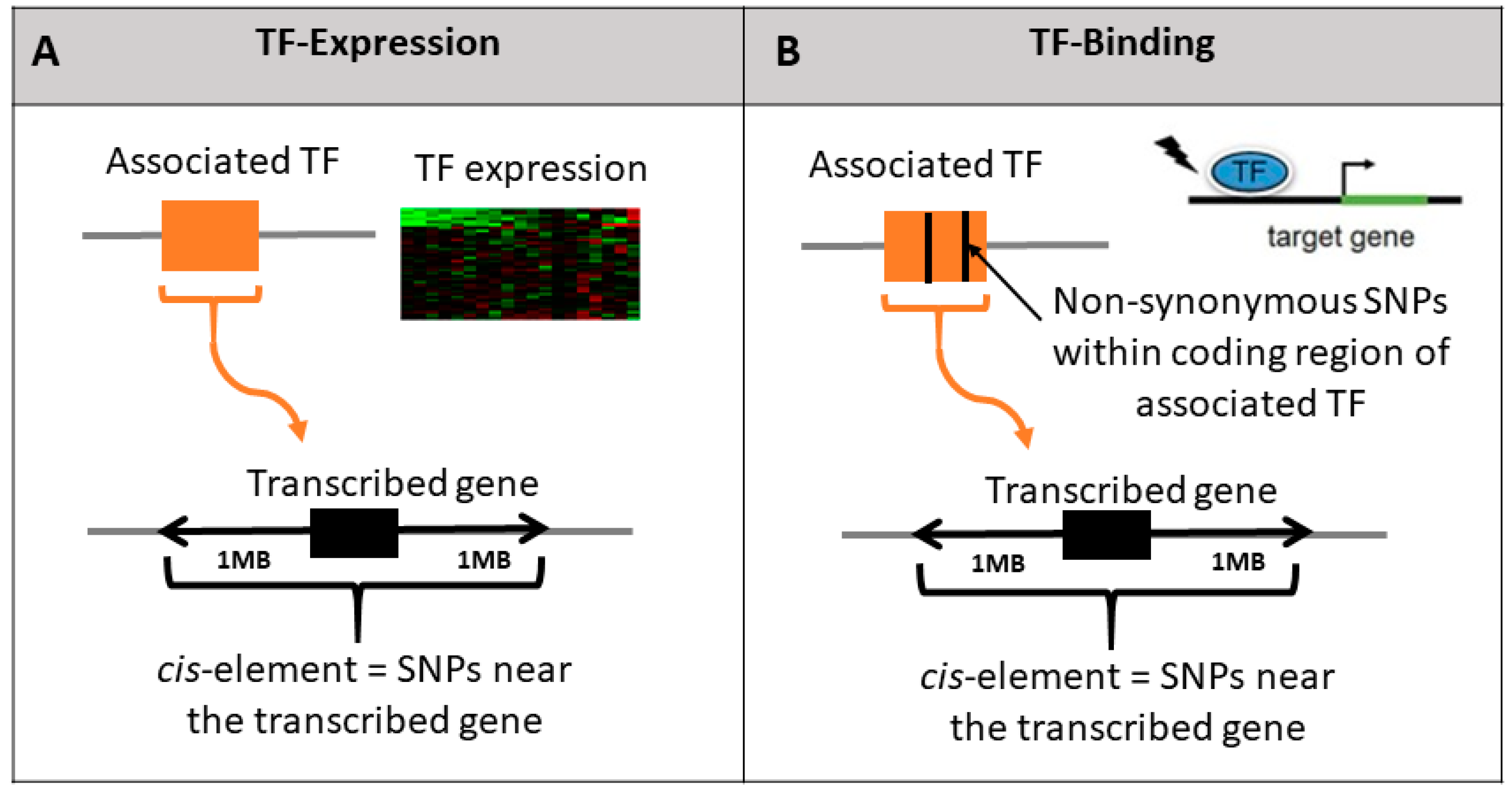
2.2. Constructing TF Models
2.3. Statistical Analysis
3. Results
3.1. Identifying Gene Expression Associated with TF Variability
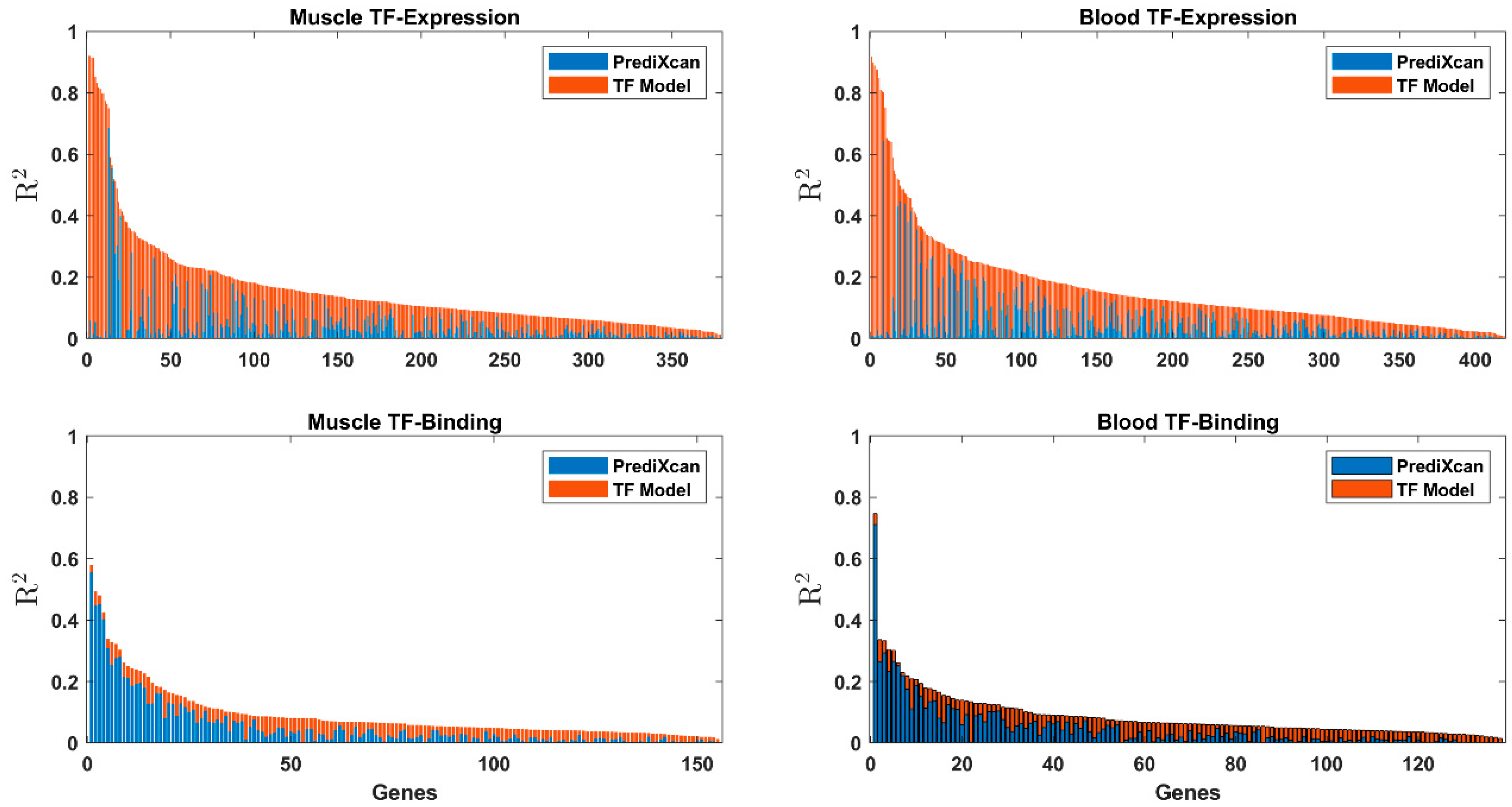
3.2. Comparison with Other Methods
3.3. Hit Genes in Expression Models Are Enriched with Immune Response
3.4. Explanatory Trans Associations Display Tissue-Specific Regulation of Immune Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nica, A.C.; Dermitzakis, E.T. Expression quantitative trait loci: Present and future. Phil. Trans. R. Soc. B 2013, 368, 20120362. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.; Jansen, R.; De Geus, E.J.; Boomsma, D.I.; Wright, F.A. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamazon, E.R.; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; Nicolae, D.L.; Cox, N.J. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, Y.I.; Pritchard, J.K. Trans effects on gene expression can drive omnigenic inheritance. Cell 2019, 177, 1022–1034.e1026. [Google Scholar] [CrossRef]
- Wheeler, H.E.; Ploch, S.; Barbeira, A.N.; Bonazzola, R.; Andaleon, A.; Fotuhi Siahpirani, A.; Saha, A.; Battle, A.; Roy, S.; Im, H.K. Imputed gene associations identify replicable trans-acting genes enriched in transcription pathways and complex traits. Genet. Epidemiol. 2019, 43, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef]
- Fujimaki, T.; Oguri, M.; Horibe, H.; Kato, K.; Matsuoka, R.; Abe, S.; Tokoro, F.; Arai, M.; Noda, T.; Watanabe, S. Association of a transcription factor 21 gene polymorphism with hypertension. Biomed. Rep. 2015, 3, 118–122. [Google Scholar] [CrossRef]
- Palizban, A.; Rezaei, M.; Khanahmad, H.; Fazilati, M. Transcription factor 7-like 2 polymorphism and context-specific risk of metabolic syndrome, type 2 diabetes, and dyslipidemia. J. Res. Med. Sci. Off. J. Isfahan Univ. Med. Sci. 2017, 22, 40. [Google Scholar] [CrossRef]
- Hamed, W.A.; Hammouda, G.E.; El-Hefnawy, S.M. Transcription factor 21 gene polymorphism in patients with coronary artery disease. Res. Rep. Clin. Cardiol. 2016, 55, 13–18. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013, 45, 580. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2017, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Bovolenta, L.A.; Acencio, M.L.; Lemke, N. HTRIdb: An open-access database for experimentally verified human transcriptional regulation interactions. BMC Genom. 2012, 13, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.-P.; Wu, C.; Miao, H.; Wu, H. RegNetwork: An integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015, 2015, bav095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Brugge, H. Large-scale cis-and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Funnell, A.P.; Norton, L.J.; Mak, K.S.; Burdach, J.; Artuz, C.M.; Twine, N.A.; Wilkins, M.R.; Power, C.A.; Hung, T.-T.; Perdomo, J. The CACCC-binding protein KLF3/BKLF represses a subset of KLF1/EKLF target genes and is required for proper erythroid maturation in vivo. Mol. Cell. Biol. 2012, 32, 3281–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Liu, K.; Sun, C.-W.; Pawlik, K.M.; Townes, T.M. KLF1 regulates BCL11A expression and γ-to β-globin gene switching. Nat. Genet. 2010, 42, 742–744. [Google Scholar] [CrossRef] [PubMed]
- Donze, D.; Townes, T.M.; Bieker, J.J. Role of Erythroid Kruppel-like Factor in Human γ-to β-Globin Gene Switching (∗). J. Biol. Chem. 1995, 270, 1955–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Zhang, H.; Lin, A.; Wu, Z.; Li, T.; Zhang, X.; Chen, H.; Lu, D. Multi-Omics Analysis in β-Thalassemia Using an HBB Gene-Knockout Human Erythroid Progenitor Cell Model. Int. J. Mol. Sci. 2022, 23, 2807. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hu, H.; Zhang, Q.; Yang, Y.; Li, Y.; Hu, Y.; Ruan, X.; Yang, Y.; Zhang, Z.; Shu, C. Dynamic transcriptomes of human myeloid leukemia cells. Genomics 2013, 102, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, Y.; Pi, W.; Liu, H.; Wickrema, A.; Tuan, D. NF-Y recruits both transcription activator and repressor to modulate tissue-and developmental stage-specific expression of human γ-globin gene. PLoS ONE 2012, e47175. [Google Scholar] [CrossRef] [Green Version]
- Doerfler, P.A.; Feng, R.; Li, Y.; Palmer, L.E.; Porter, S.N.; Bell, H.W.; Crossley, M.; Pruett-Miller, S.M.; Cheng, Y.; Weiss, M.J. Activation of γ-globin gene expression by GATA1 and NF-Y in hereditary persistence of fetal hemoglobin. Nat. Genet. 2021, 53, 1177–1186. [Google Scholar] [CrossRef]
- Gee, B.E.; Pearson, A.; Buchanan-Perry, I.; Simon, R.P.; Archer, D.R.; Meller, R. Whole Blood Transcriptome Analysis in Children with Sickle Cell Anemia. Front. Genet. 2021, 12, 737741. [Google Scholar] [CrossRef]
- Wakil, S.M.; Alhissi, S.; Al Dossari, H.; Alqahtani, A.; Shibin, S.; Melaiki, B.T.; Finsterer, J.; Al-Hashem, A.; Bohlega, S.; Alazami, A.M. Truncating ARL6IP1 variant as the genetic cause of fatal complicated hereditary spastic paraplegia. BMC Med Genet. 2019, 20, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Nizon, M.; Küry, S.; Péréon, Y.; Besnard, T.; Quinquis, D.; Boisseau, P.; Marsaud, T.; Magot, A.; Mussini, J.M.; Mayrargue, E. ARL6IP1 mutation causes congenital insensitivity to pain, acromutilation and spastic paraplegia. Clin. Genet. 2018, 93, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Di Lisi, R.; Picard, A.; Ausoni, S.; Schiaffino, S. GATA elements control repression of cardiac troponin I promoter activity in skeletal muscle cells. BMC Mol. Biol. 2007, 8, 1–8. [Google Scholar]
- Musarò, A.; McCullagh, K.J.; Naya, F.J.; Olson, E.N.; Rosenthal, N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature 1999, 400, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.C.; Rosenthal, N. Different modes of hypertrophy in skeletal muscle fibers. J. Cell Biol. 2002, 156, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Lin, J.; Hsu, C.; Juan, H.; Lou, P.; Huang, M. GATA3 interacts with and stabilizes HIF-1α to enhance cancer cell invasiveness. Oncogene 2017, 36, 4243–4252. [Google Scholar] [CrossRef]
- Remels, A.; Langen, R.; Gosker, H.R.; Russell, A.; Spaapen, F.; Voncken, J.; Schrauwen, P.; Schols, A.M. PPARγ inhibits NF-κB-dependent transcriptional activation in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E174–E183. [Google Scholar] [CrossRef]
- Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An aPPARent functional consequence in skeletal muscle physiology via peroxisome proliferator-activated receptors. Int. J. Mol. Sci. 2018, 19, 1425. [Google Scholar] [CrossRef] [Green Version]
- De Rooij, J.D.; Beuling, E.; van den Heuvel-Eibrink, M.M.; Obulkasim, A.; Baruchel, A.; Trka, J.; Reinhardt, D.; Sonneveld, E.; Gibson, B.E.; Pieters, R. Recurrent deletions of IKZF1 in pediatric acute myeloid leukemia. Haematologica 2015, 100, 1151. [Google Scholar] [CrossRef] [Green Version]
- Leng, D.; Miao, R.; Huang, X.; Wang, Y. In silico analysis identifies CRISP3 as a potential peripheral blood biomarker for multiple myeloma: From data modeling to validation with RT-PCR. Oncol. Lett. 2018, 15, 5167–5174. [Google Scholar] [CrossRef] [Green Version]
- Pfisterer, P.; König, H.; Hess, J.; Lipowsky, G.; Haendler, B.; Schleuning, W.-D.; Wirth, T. CRISP-3, a protein with homology to plant defense proteins, is expressed in mouse B cells under the control of Oct2. Mol. Cell. Biol. 1996, 16, 6160–6168. [Google Scholar] [CrossRef] [Green Version]
- Ng, A.P.; Coughlan, H.D.; Hediyeh-Zadeh, S.; Behrens, K.; Johanson, T.M.; Low, M.S.Y.; Bell, C.C.; Gilan, O.; Chan, Y.-C.; Kueh, A.J. An Erg-driven transcriptional program controls B cell lymphopoiesis. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Knief, J.; Reddemann, K.; Gliemroth, J.; Brede, S.; Bartscht, T.; Thorns, C. ERG expression in multiple myeloma—A potential diagnostic pitfall. Pathol. Res. Pract. 2017, 213, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S.; Taguchi, O.; Seto, M. Promotion and maintenance of leukemia by ERG. Blood J. Am. Soc. Hematol. 2011, 117, 3858–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczera, P.; Martin, L.; Marx, G.; Schuerholz, T. The ribonuclease a superfamily in humans: Canonical RNases as the buttress of innate immunity. Int. J. Mol. Sci. 2016, 17, 1278. [Google Scholar] [CrossRef] [PubMed]
- Parakati, R.; DiMario, J.X. Sp1-and Sp3-mediated transcriptional regulation of the fibroblast growth factor receptor 1 gene in chicken skeletal muscle cells. J. Biol. Chem. 2002, 277, 9278–9285. [Google Scholar] [CrossRef] [Green Version]
- Irrcher, I.; Hood, D.A. Regulation of Egr-1, SRF, and Sp1 mRNA expression in contracting skeletal muscle cells. J. Appl. Physiol. 2004, 97, 2207–2213. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes-Diaz, C.; Delaporte, C.; Dautréaux, B.; Charron, D.; Fardeau, M. Class II MHC antigens in normal human skeletal muscle. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 1992, 15, 295–302. [Google Scholar] [CrossRef]
- Englund, P.; Lindroos, E.; Nennesmo, I.; Klareskog, L.; Lundberg, I.E. Skeletal muscle fibers express major histocompatibility complex class II antigens independently of inflammatory infiltrates in inflammatory myopathies. Am. J. Pathol. 2001, 159, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Van Den Elsen, P.J. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front. Immunol. 2011, 2, 48. [Google Scholar] [CrossRef] [Green Version]
- Castelli, E.C.; Veiga-Castelli, L.C.; Yaghi, L.; Moreau, P.; Donadi, E.A. Transcriptional and posttranscriptional regulations of the HLA-G gene. J. Immunol. Res. 2014, 2014, 734068. [Google Scholar] [CrossRef] [Green Version]
- Carli, L.; Tani, C.; Vagnani, S.; Signorini, V.; Mosca, M. Leukopenia, lymphopenia, and neutropenia in systemic lupus erythematosus: Prevalence and clinical impact—A systematic literature review. In Seminars in Arthritis and Rheumatism; Elsevier: Amsterdam, The Netherlands, 2015; Volume 45, pp. 190–194. [Google Scholar]
- Bitencourt, N.; Solow, E.B.; Wright, T.; Bermas, B.L. Inflammatory myositis in systemic lupus erythematosus. Lupus 2020, 29, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.M.; Keenan, K.; Senn, N.; Hutcheon, G.; Chan, K.; Erwig, L.; Schrepf, A.; Dospinescu, P.; Gray, S.; Waiter, G. Metabolic and Structural Skeletal Muscle Health in Systemic Lupus Erythematosus–Related Fatigue: A Multimodal Magnetic Resonance Imaging Study. Arthritis Care Res. 2019, 71, 1640–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vechetti, I.J., Jr.; Valentino, T.; Mobley, C.B.; McCarthy, J.J. The role of extracellular vesicles in skeletal muscle and systematic adaptation to exercise. J. Physiol. 2021, 599, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Rome, S.; Forterre, A.; Mizgier, M.L.; Bouzakri, K. Skeletal muscle-released extracellular vesicles: State of the art. Front. Physiol. 2019, 929. [Google Scholar] [CrossRef]
- Lu, Z.; Joseph, D.; Bugnard, E.; Zaal, K.J.; Ralston, E. Golgi complex reorganization during muscle differentiation: Visualization in living cells and mechanism. Mol. Biol. Cell 2001, 12, 795–808. [Google Scholar] [CrossRef] [Green Version]
- Ralston, E.; Ploug, T.; Kalhovde, J.; Lømo, T. Golgi complex, endoplasmic reticulum exit sites, and microtubules in skeletal muscle fibers are organized by patterned activity. J. Neurosci. 2001, 21, 875–883. [Google Scholar] [CrossRef]
- Donkervoort, S.; Krause, N.; Dergai, M.; Yun, P.; Koliwer, J.; Gorokhova, S.; Geist Hauserman, J.; Cummings, B.B.; Hu, Y.; Smith, R. BET1 variants establish impaired vesicular transport as a cause for muscular dystrophy with epilepsy. EMBO Mol. Med. 2021, 13, e13787. [Google Scholar] [CrossRef]
- Buas, M.F.; Kadesch, T. Regulation of skeletal myogenesis by Notch. Exp. Cell Res. 2010, 316, 3028–3033. [Google Scholar] [CrossRef] [Green Version]
- Raj, P.; Rai, E.; Song, R.; Khan, S.; Wakeland, B.E.; Viswanathan, K.; Arana, C.; Liang, C.; Zhang, B.; Dozmorov, I. Regulatory polymorphisms modulate the expression of HLA class II molecules and promote autoimmunity. eLife 2016, 5, e12089. [Google Scholar] [CrossRef]
- Carey, B.S.; Poulton, K.V.; Poles, A. Factors affecting HLA expression: A review. Int. J. Immunogenet. 2019, 46, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Bronson, P.G.; Caillier, S.; Ramsay, P.P.; McCauley, J.L.; Zuvich, R.L.; De Jager, P.L.; Rioux, J.D.; Ivinson, A.J.; Compston, A.; Hafler, D.A. CIITA variation in the presence of HLA-DRB1* 1501 increases risk for multiple sclerosis. Hum. Mol. Genet. 2010, 19, 2331–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyllenberg, A.; Piehl, F.; Alfredsson, L.; Hillert, J.; Bomfim, I.L.; Padyukov, L.; Orho-Melander, M.; Lindholm, E.; Landin-Olsson, M.; Lernmark, Å. Variability in the CIITA gene interacts with HLA in multiple sclerosis. Genes Immun. 2014, 15, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyllenberg, A.; Asad, S.; Piehl, F.; Swanberg, M.; Padyukov, L.; Van Yserloo, B.; Rutledge, E.A.; McNeney, B.; Graham, J.; Orho-Melander, M. Age-dependent variation of genotypes in MHC II transactivator gene (CIITA) in controls and association to type 1 diabetes. Genes Immun. 2012, 13, 632–640. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.; Varney, M.D.; Harrison, L.C.; Morahan, G. Definition of high-risk type 1 diabetes HLA-DR and HLA-DQ types using only three single nucleotide polymorphisms. Diabetes 2013, 62, 2135–2140. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.; Muller, Y.; Hanson, R.; Knowler, W.; Mason, C.; Bian, L.; Ossowski, V.; Wiedrich, K.; Chen, Y.; Marcovina, S. HLA-DRB1 reduces the risk of type 2 diabetes mellitus by increased insulin secretion. Diabetologia 2011, 54, 1684–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinniah, R.; Sevak, V.; Pandi, S.; Ravi, P.M.; Vijayan, M.; Kannan, A.; Karuppiah, B. HLA-DRB1 genes and the expression dynamics of HLA CIITA determine the susceptibility to T2DM. Immunogenetics 2021, 73, 291–305. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Bouzakri, K.; Koistinen, H.A.; Zierath, J.R. Molecular mechanisms of skeletal muscle insulin resistance in type 2 diabetes. Curr. Diabetes Rev. 2005, 1, 167–174. [Google Scholar] [CrossRef]
- Israni, N.; Goswami, R.; Kumar, A.; Rani, R. Interaction of vitamin D receptor with HLA DRB1* 0301 in type 1 diabetes patients from North India. PLoS ONE 2009, 4, e8023. [Google Scholar] [CrossRef] [Green Version]
- Cramer, L.A.; Nelson, S.L.; Klemsz, M.J. Synergistic induction of the Tap-1 gene by IFN-γ and lipopolysaccharide in macrophages is regulated by STAT1. J. Immunol. 2000, 165, 3190–3197. [Google Scholar] [CrossRef] [Green Version]
- White, L.C.; Wright, K.L.; Felix, N.J.; Ruffner, H.; Reis, L.F.; Pine, R.; Ting, J.P.-Y. Regulation of LMP2 and TAP1 genes by IRF-1 explains the paucity of CD8+ T cells in IRF-1−/− mice. Immunity 1996, 5, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. IRF and STAT transcription factors-from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front. Immunol. 2019, 9, 3047. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Grandvaux, N. STAT2 and IRF9: Beyond ISGF3. Jak-Stat 2013, 2, e27521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rengachari, S.; Groiss, S.; Devos, J.M.; Caron, E.; Grandvaux, N.; Panne, D. Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function. Proc. Natl. Acad. Sci. USA 2018, 115, E601–E609. [Google Scholar] [CrossRef] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Hemann, E.A.; Gale, M., Jr.; Savan, R. Interferon lambda genetics and biology in regulation of viral control. Front. Immunol. 2017, 8, 1707. [Google Scholar] [CrossRef]
- Rivera, A. Interferon lambda’s new role as regulator of neutrophil function. J. Interferon Cytokine Res. 2019, 39, 609–617. [Google Scholar] [CrossRef]
- Paul, A.; Tang, T.H.; Ng, S.K. Interferon regulatory factor 9 structure and regulation. Front. Immunol. 2018, 9, 1831. [Google Scholar] [CrossRef] [Green Version]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [Green Version]
- Blaszczyk, K.; Olejnik, A.; Nowicka, H.; Ozgyin, L.; Chen, Y.-L.; Chmielewski, S.; Kostyrko, K.; Wesoly, J.; Balint, B.L.; Lee, C.-K. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem. J. 2015, 466, 511. [Google Scholar] [CrossRef] [Green Version]
- Thibault, D.L.; Chu, A.D.; Graham, K.L.; Balboni, I.; Lee, L.Y.; Kohlmoos, C.; Landrigan, A.; Higgins, J.P.; Tibshirani, R.; Utz, P.J. IRF9 and STAT1 are required for IgG autoantibody production and B cell expression of TLR7 in mice. J. Clin. Investig. 2008, 118, 1417–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goropevšek, A.; Gorenjak, M.; Gradišnik, S.; Dai, K.; Holc, I.; Hojs, R.; Krajnc, I.; Pahor, A.; Avčin, T. Increased levels of STAT1 protein in blood CD4 T cells from systemic lupus erythematosus patients are associated with perturbed homeostasis of activated CD45RA-FOXP3hi regulatory subset and follow-up disease severity. J. Interferon Cytokine Res. 2017, 37, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dai, M.; Cui, Y.; Hou, G.; Deng, J.; Gao, X.; Liao, Z.; Liu, Y.; Meng, Y.; Wu, L. Association of Abnormal Elevations in IFIT 3 With Overactive Cyclic GMP-AMP Synthase/Stimulator of Interferon Genes Signaling in Human Systemic Lupus Erythematosus Monocytes. Arthritis Rheumatol. 2018, 70, 2036–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Li, M.; Wang, Z.; Wang, J. Ifn-γ mediates the development of systemic lupus erythematosus. BioMed Res. Int. 2020, 2020, 7176515. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gamazon, E.R.; Pierce, B.L.; Stranger, B.E.; Im, H.K.; Gibbons, R.D.; Cox, N.J.; Nicolae, D.L.; Chen, L.S. Imputing gene expression in uncollected tissues within and beyond GTEx. Am. J. Hum. Genet. 2016, 98, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Ramos, C.M.; Paulson, J.N.; Chen, C.-Y.; Kuijjer, M.L.; Fagny, M.; Platig, J.; Sonawane, A.R.; DeMeo, D.L.; Quackenbush, J.; Glass, K. Regulatory network changes between cell lines and their tissues of origin. BMC Genom. 2017, 18, 723. [Google Scholar] [CrossRef] [Green Version]
- Devenish, L.P.; Mhlanga, M.M.; Negishi, Y. Immune Regulation in Time and Space: The Role of Local-and Long-Range Genomic Interactions in Regulating Immune Responses. Front. Immunol. 2021, 12, 1715. [Google Scholar] [CrossRef]
- Smale, S.T. Transcriptional regulation in the immune system: A status report. Trends Immunol. 2014, 35, 190–194. [Google Scholar] [CrossRef] [Green Version]
- helu2008/transTFModel. Available online: https://github.com/helu2008/transTFModel (accessed on 4 August 2021).




{kind=link}
{kind=link}
{kind=link}
| Skeletal Muscle | Whole Blood | |
|---|---|---|
| Number of samples | 706 | 670 |
| Number of expressed genes | 21,031 | 20,315 |
| Number of expressed genes with an associated TF (% of genes) | 11,130 (53%) | 10,563 (52%) |
| Number TFs per gene | 10.8 ± 8.4 | 10.9 ± 8.7 |
| Number of nsSNP * per TF | 1.55 ± 1.77 | 1.57 ± 1.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Tang, Y.-C.; Gottlieb, A. Tissue-Specific Variations in Transcription Factors Elucidate Complex Immune System Regulation. Genes 2022, 13, 929. https://doi.org/10.3390/genes13050929
Lu H, Tang Y-C, Gottlieb A. Tissue-Specific Variations in Transcription Factors Elucidate Complex Immune System Regulation. Genes. 2022; 13(5):929. https://doi.org/10.3390/genes13050929
Chicago/Turabian StyleLu, Hengwei, Yi-Ching Tang, and Assaf Gottlieb. 2022. "Tissue-Specific Variations in Transcription Factors Elucidate Complex Immune System Regulation" Genes 13, no. 5: 929. https://doi.org/10.3390/genes13050929
APA StyleLu, H., Tang, Y. -C., & Gottlieb, A. (2022). Tissue-Specific Variations in Transcription Factors Elucidate Complex Immune System Regulation. Genes, 13(5), 929. https://doi.org/10.3390/genes13050929