
Identification of the Key miRNAs and Genes Associated with the Regulation of Non-Small Cell Lung Cancer: A Network-Based Approach

 , , , and
, , , and
Abstract
:1. Introduction
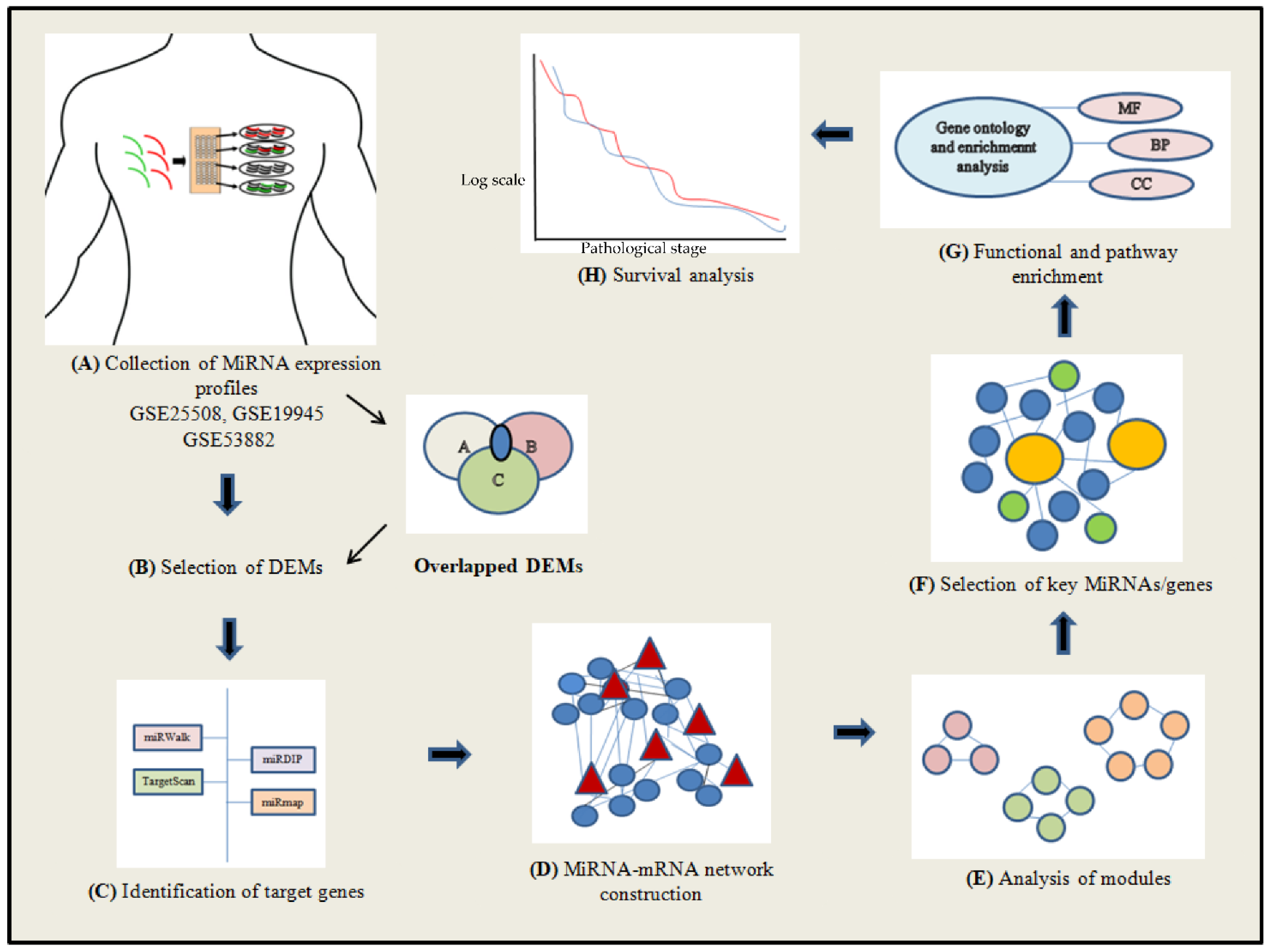
2. Materials and Methods
2.1. Search Strategy and Inclusion Criteria of Studies
2.2. Acquisition of MiRNA Expression Data
2.3. Data Preprocessing and Screening of Differentially Expressed MiRNAs (DEMs)
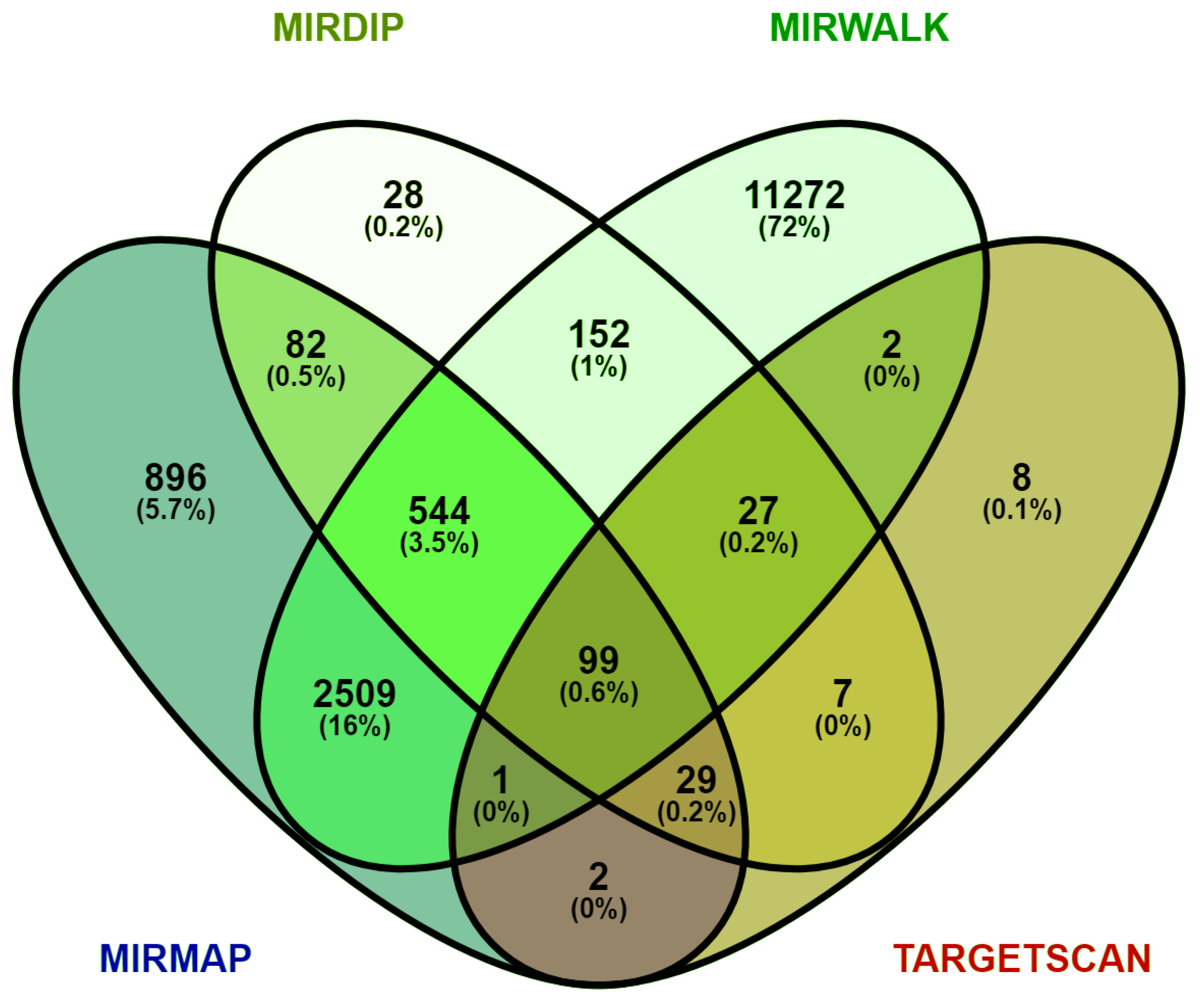
2.4. Identification of the DEM Target Genes
2.5. DEM–mRNA Network Construction
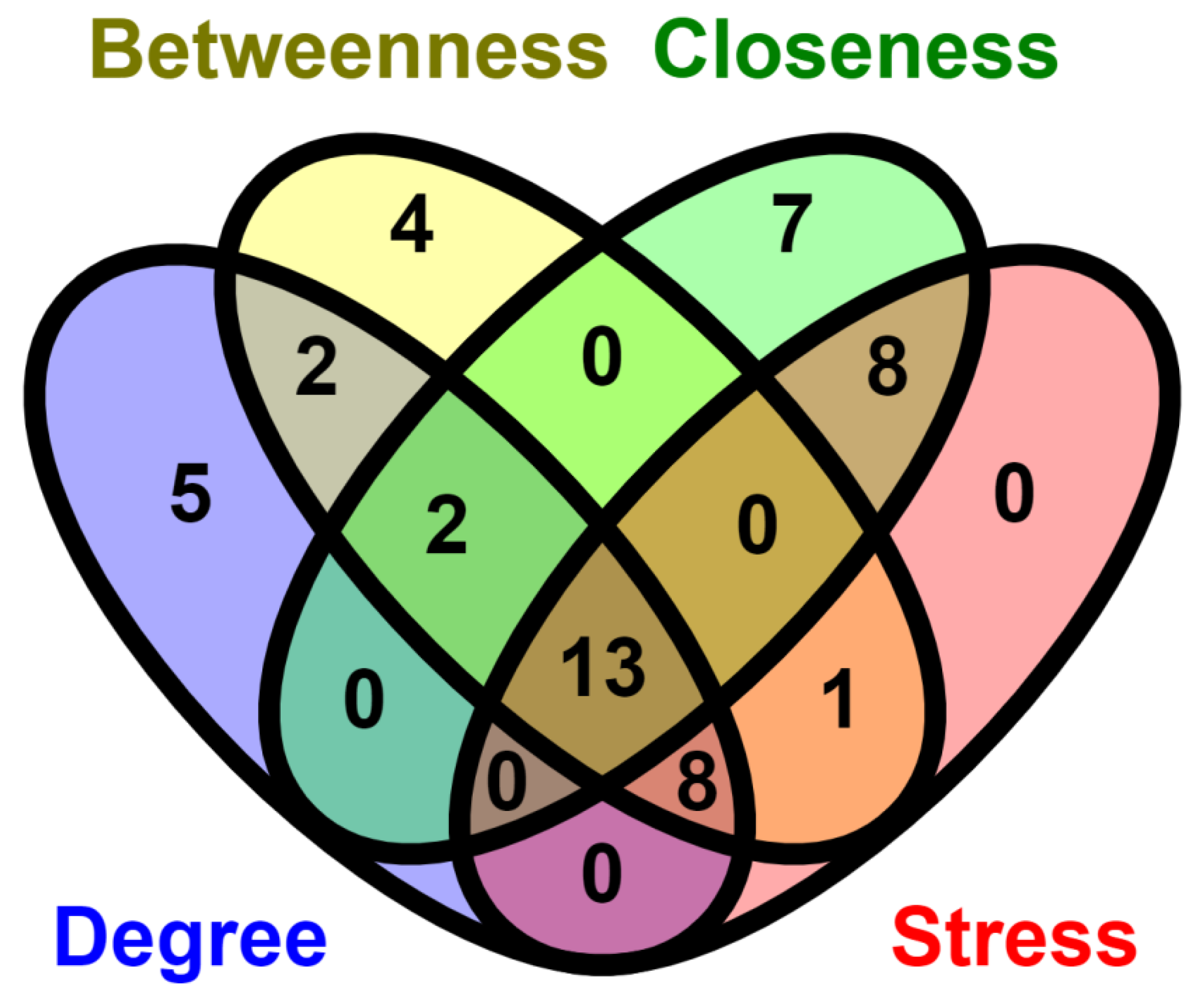
2.6. Network Analysis
2.7. Gene Term Enrichment and Pathway Analysis
2.8. Survival Analysis and Prediction of Transcription Factors (TFs)
3. Results
3.1. Selection of DEGs
3.2. Selection of MiRNA from Datasets
3.3. Prediction of Target Genes for DEMs
3.4. Construction of the MiRNA–mRNA Network
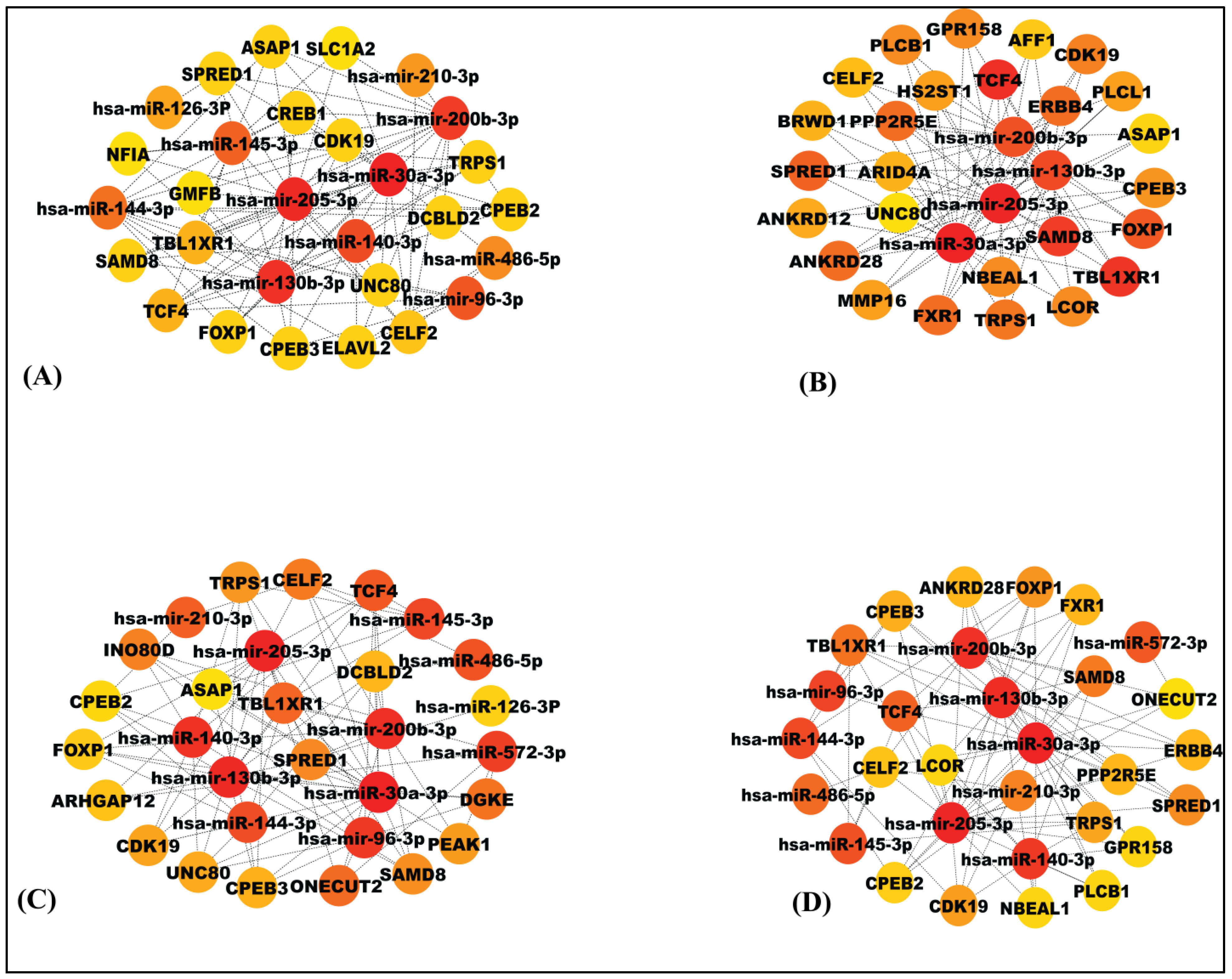
3.5. Detection of Significant Modules
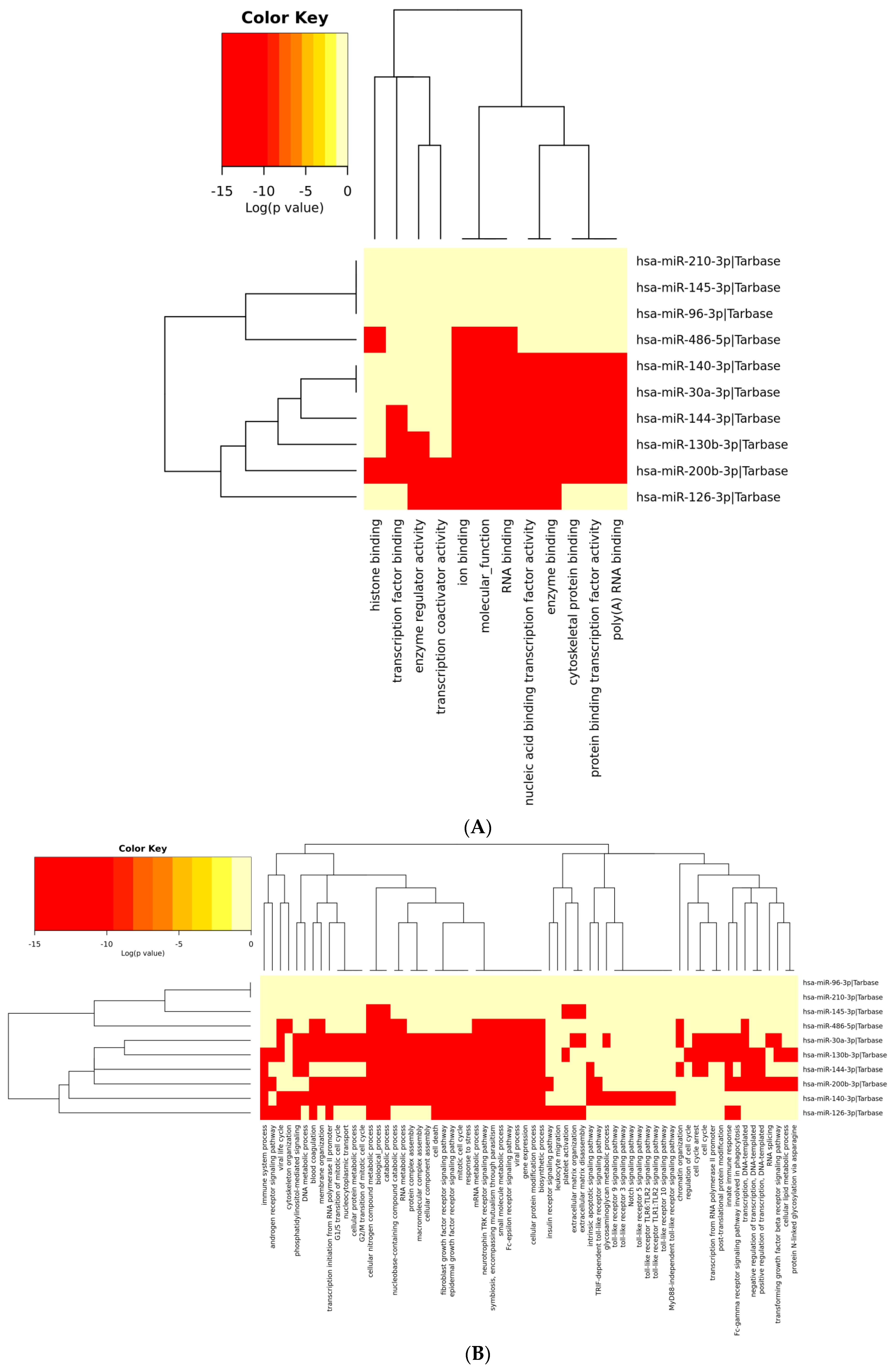
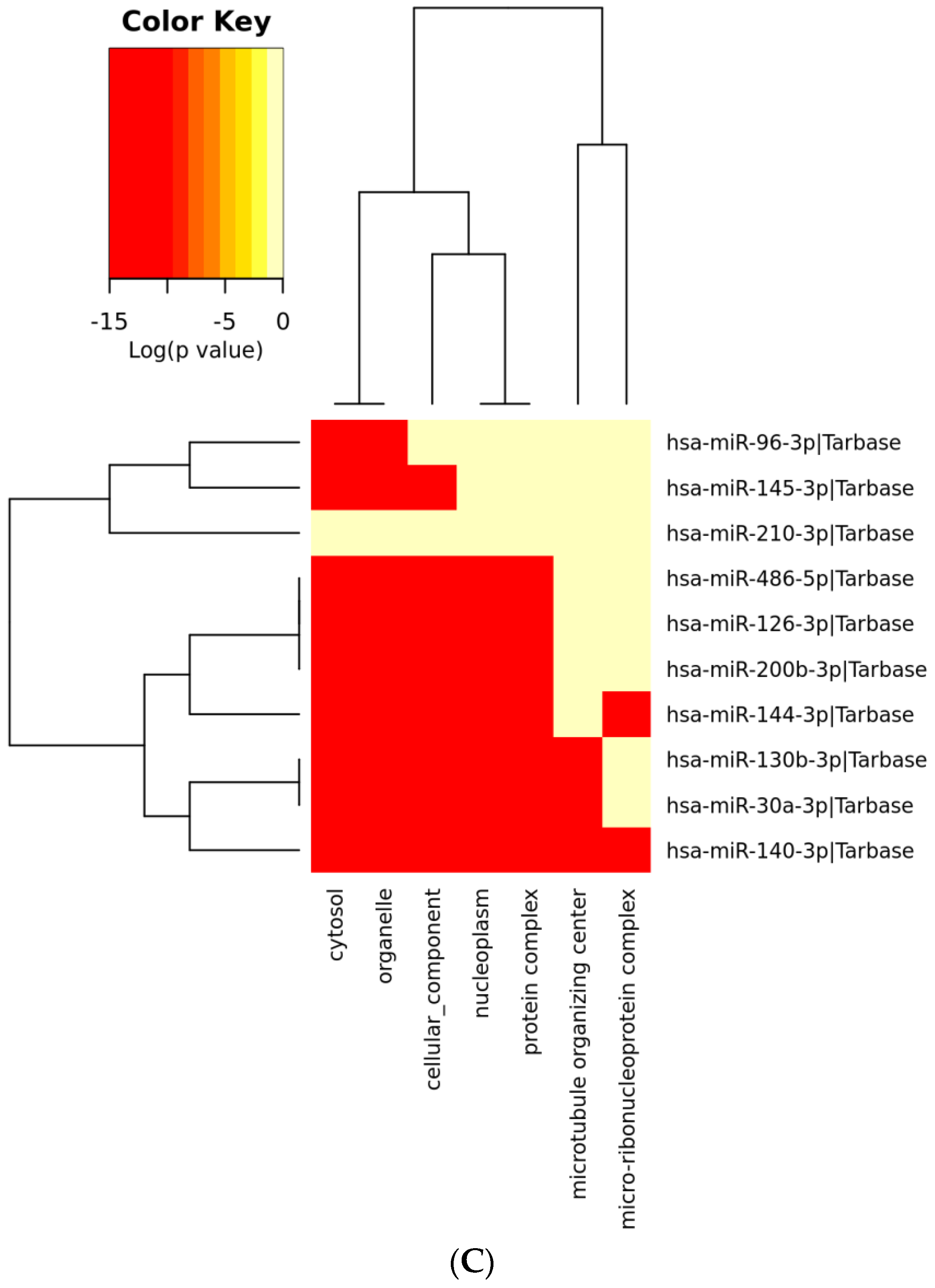
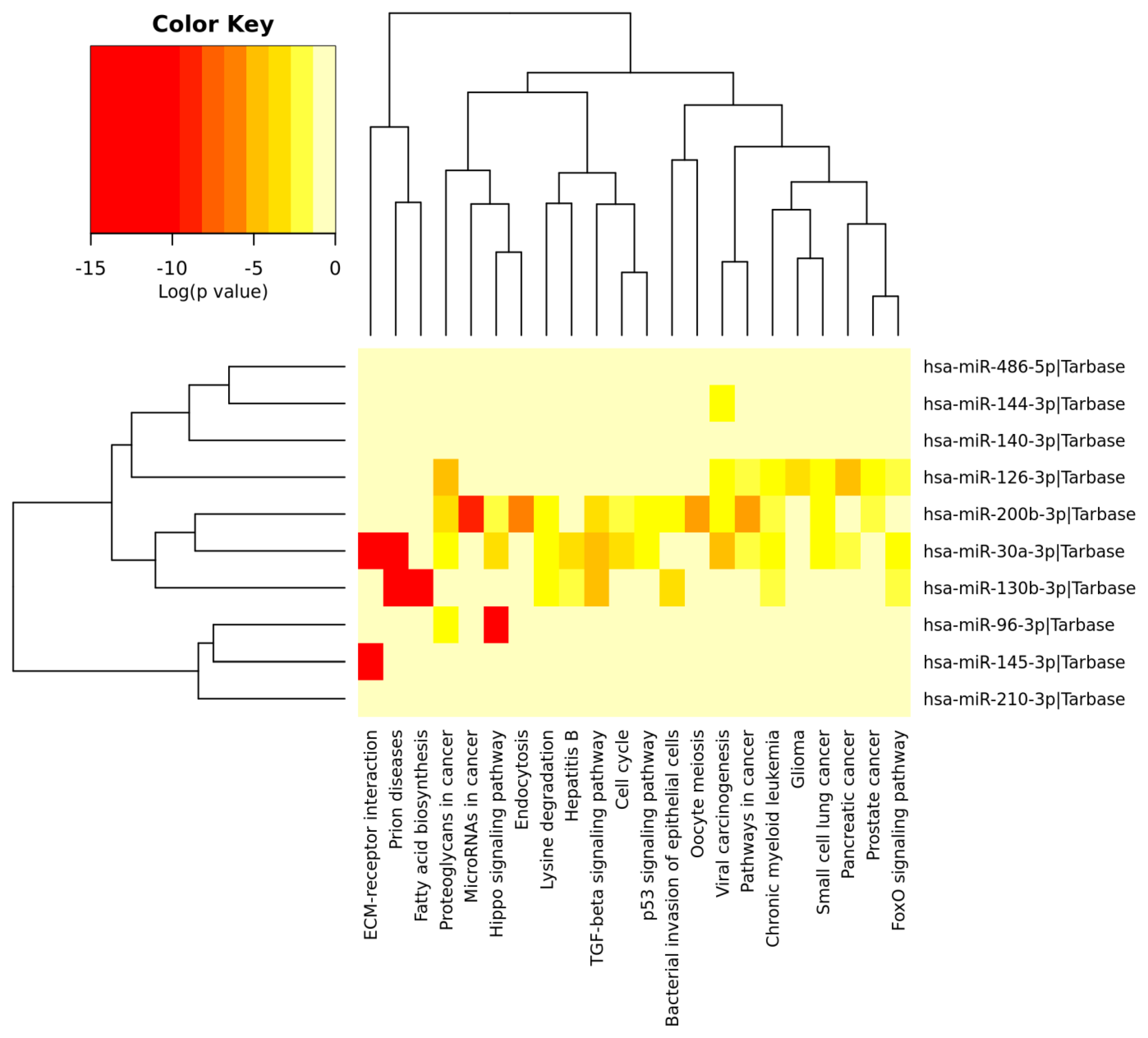
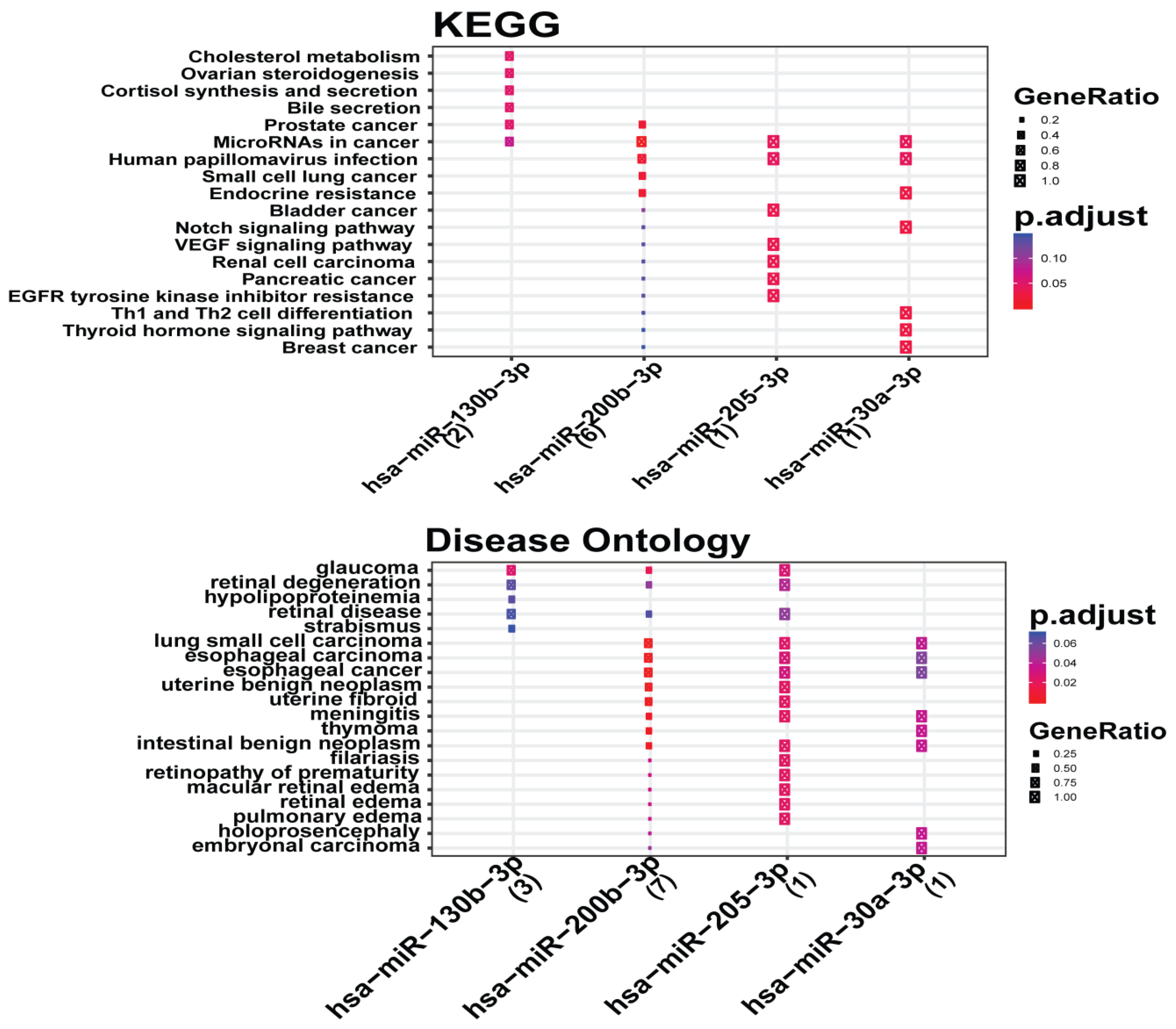
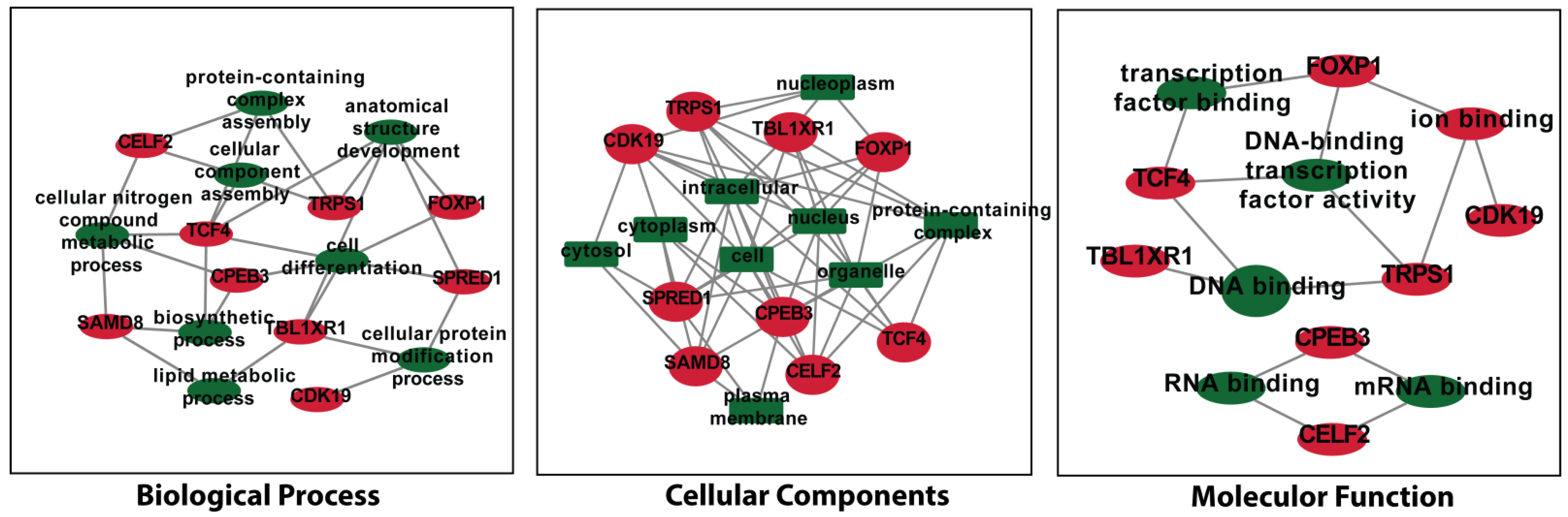
3.6. Analysis of Gene Term Enrichment and Pathways
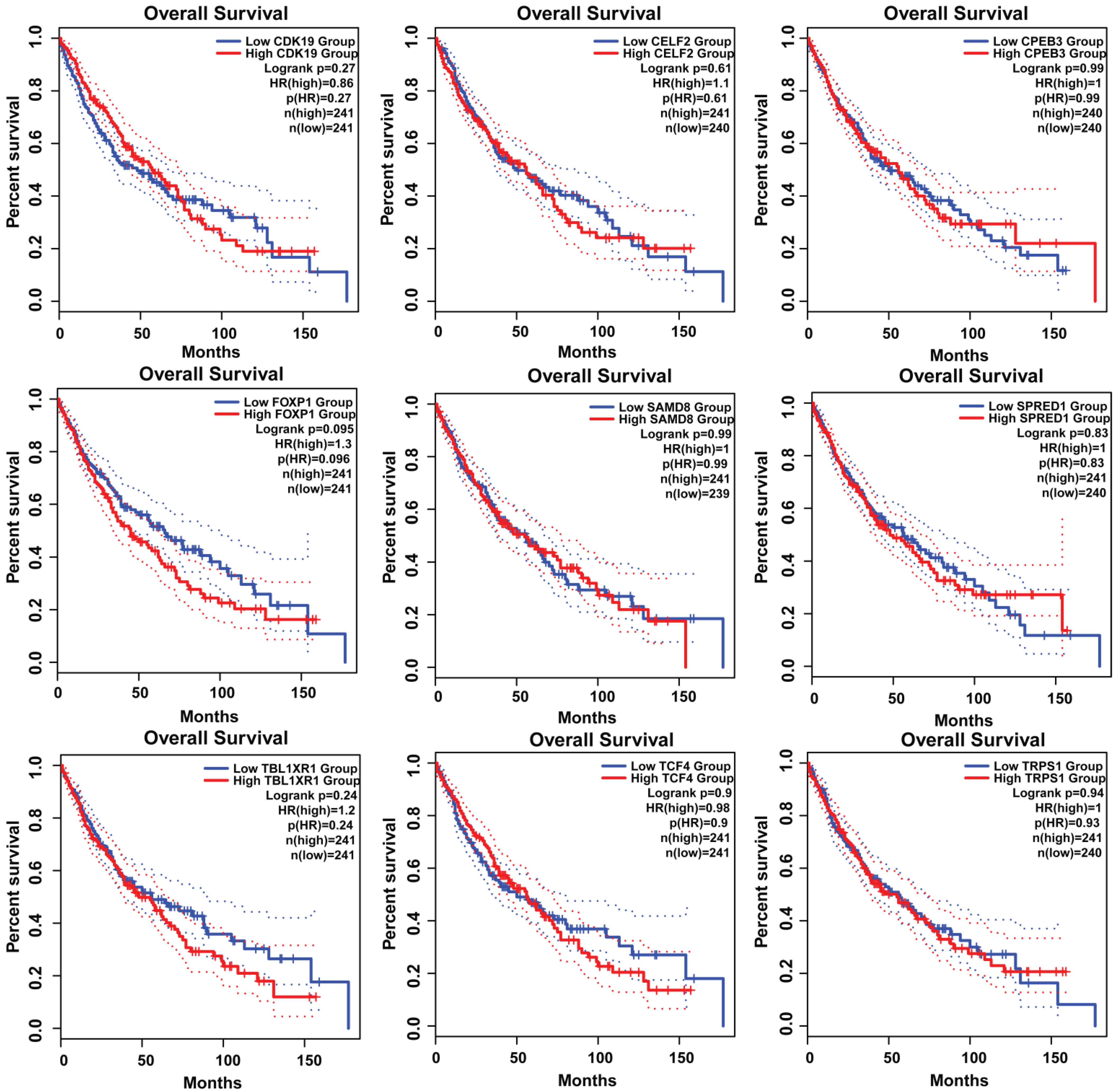
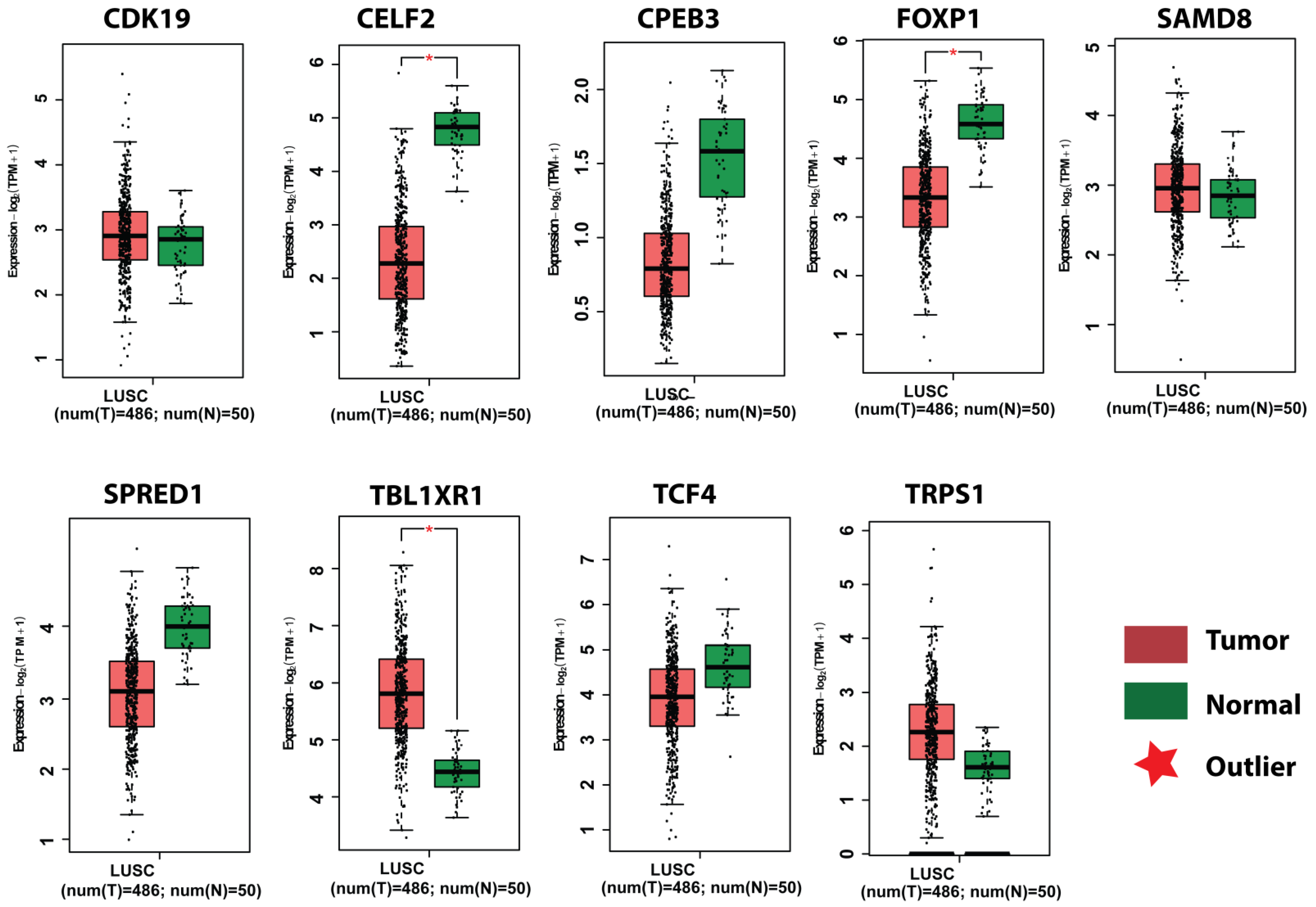
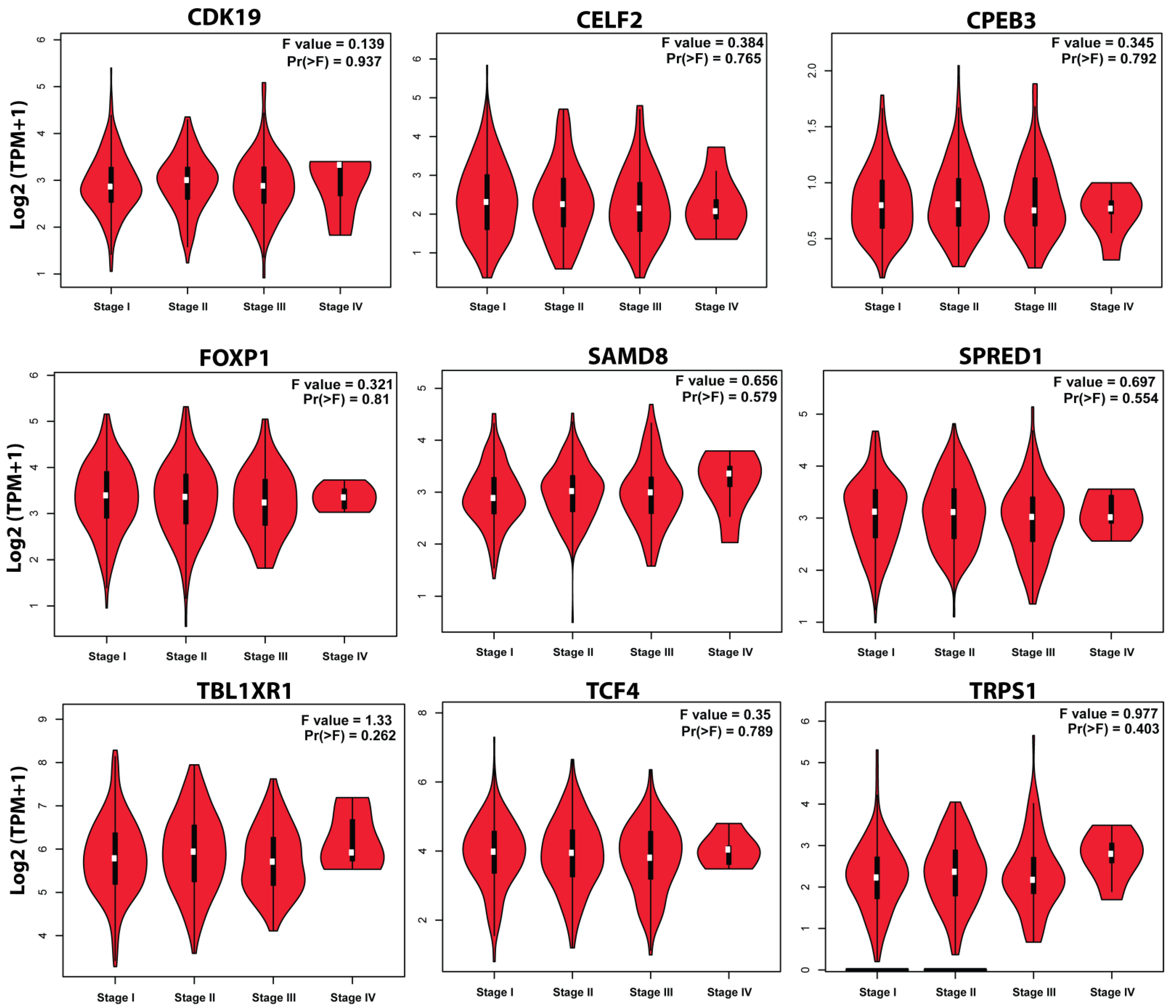
3.7. Survival Plot Analysis of the Key Genes
3.8. Identification of the TFs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CAA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Cancer Society. “Non-Small Cell Lung Cancer”. May 2018. Available online: https://www.cancer.org/cancer/non-small-celllung-cancer.html (accessed on 1 June 2021).
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Perez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): Arandomised controlled trial. Lancet 2015, 387, 1540–1550. [Google Scholar] [CrossRef]
- Fumarola, C.; Bonelli, M.A.; Petronini, P.G.; Alfieri, R.R. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem. Pharmacol. 2014, 90, 197–207. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. ROS1 protein-tyrosine kinase inhibitors in the treatment of ROS1 fusion protein-driven non-small cell lung cancers. Pharmacol. Res. 2017, 121, 202–212. [Google Scholar] [CrossRef]
- Best, S.A.; De Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab. 2018, 27, 935–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, N.; Johnson, D.; Temin, S.; Baker, S., Jr.; Brahmer, J.; Ellis, P.M.; Giaccone, G.; Hesketh, P.J.; Jaiyesimi, I.; Leighl, N.B.; et al. Systemic therapy for stage IV non–small-cell lung cancer: American Society of Clinical Oncology clinical practice guideline update. J. Clin. Oncol. 2017, 35, 3484–3515. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Ishikawa, Y. MicroRNA In Lung Cancer: Novel Biomarkers and Potential Tools for Treatment. J. Clin. Med. 2016, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Inamura, K. Diagnostic and Therapeutic Potential of MicroRNAs in Lung Cancer. Cancers 2017, 9, 49. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cobb, G.; Anderson, T. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Guan, P.; Yin, Z.; Li, X.; Wu, W.; Zhou, B. Meta-analysis of human lung cancer microRNA expression profiling studies comparing cancer tissues with normal tissues. J. Exp. Clin. Cancer Res. 2012, 31, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Zhan, B.; Lu, D.; Luo, P.; Wang, B. Prognostic Value of Expression of MicroRNAs in Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Clin. Lab. 2016, 62, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Berghmans, T.; Ameye, L.; Willems, L.; Paesmans, M.; Mascaux, C.; Lafitte, J.J.; Meert, A.P.; Scherpereel, A.; Cortot, A.B.; CsToth, I.; et al. Identification of microRNA-based signatures for response and survival for non-small cell lung cancer treated with cisplatin-vinorelbine A ELCWP prospective study. Lung Cancer 2013, 82, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Markou, A.; Sourvinou, I.; Vorkas, P.; Yousef, G.; Lianidou, E. Clinical evaluation of microRNA expression profiling in non small cell lung cancer. Lung Cancer 2013, 81, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Kalari, K.R.; Rossell, D.; Necela, B.M.; Asmann, Y.W.; Nair, A.; Baheti, S.; Kachergus, J.M.; Younkin, C.S.; Baker, T.R.; Carr, J.M.; et al. Deep Sequence Analysis of Non-Small Cell Lung Cancer: Integrated Analysis of Gene Expression, Alternative Splicing, and Single Nucleotide Variations in Lung Adenocarcinomas with and without Oncogenic KRAS Mutations. Front. Oncol. 2012, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.-L.; Wang, H.; Liu, J.; Wang, Z.-X. MicroRNA-21 (miR-21) expression promotes growth, metastasis, and chemo- or radioresistance in non-small cell lung cancer cells by targeting PTEN. Mol. Cell. Biochem. 2012, 372, 35–45. [Google Scholar] [CrossRef]
- Võsa, U.; Vooder, T.; Kolde, R.; Vilo, J.; Metspalu, A.; Annilo, T. Meta-analysis of microRNA expression in lung cancer. Int. J. Cancer 2012, 132, 2884–2893. [Google Scholar] [CrossRef]
- Cui, R.; Meng, W.; Sun, H.-L.; Kim, T.; Ye, Z.; Fassan, M.; Jeon, Y.-J.; Li, B.; Vicentini, C.; Peng, Y.; et al. MicroRNA-224 promotes tumor progression in nonsmall cell lung cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4288–E4297. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Guo, W.; Qiu, Z.; Wang, Q.; Li, Y.; Liang, L.; Liu, L.; Huang, S.; Zhao, Y.; He, X. MicroRNA-30d-5p inhibits tumour cell proliferation and motility by directly targeting CCNE2 in non-small cell lung cancer. Cancer Lett. 2015, 362, 208–217. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar]
- Lewis, B.P.; Shih, I.-H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Vejnar, C.; Zdobnov, E.M. miRmap: Comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk–Database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.-C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1—Integrative database of human microRNA target predictions. Nucleic Acids Res. 2017, 46, D360–D370. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, A.; Keskin, O.; Nussinov, R. Topological properties of protein interaction networks from a structural perspective. Biochem. Soc. Trans. 2008, 36, 1398–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, K. Construction and analysis of protein–protein interaction networks. Autom. Exp. 2010, 2, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Brandes, U. A faster algorithm for betweenness centrality. J. Math. Sociol. 2001, 25, 163–177. [Google Scholar] [CrossRef]
- Albert, R.; Barabási, A.-L. Statistical mechanics of complex networks. Rev. Mod. Phys. 2002, 74, 47–97. [Google Scholar] [CrossRef] [Green Version]
- Maslov, S.; Sneppen, K. Specificity and Stability in Topology of Protein Networks. Science 2002, 296, 910–913. [Google Scholar] [CrossRef] [Green Version]
- Canright, G.; Engø-Monsen, K. Roles in networks. Sci. Comput. Program. 2004, 53, 195–214. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, K.A.; Kaufmann, M. Decentralized Algorithms for Evaluating Centrality in Complex Networks; Universitätsbibliothek Tübingen: Tübingen, Germany, 2003. [Google Scholar]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An interactive web tool for microRNA-target enrichment and network-based analysis. BMC Bioinform. 2019, 20, 545. [Google Scholar] [CrossRef] [Green Version]
- Võsa, U.; Kolde, R.; Vilo, J.; Metspalu, A.; Annilo, T. Comprehensive Meta-analysis of MicroRNA Expression Using a Robust Rank Aggregation Approach. Methods Mol. Biol. 2014, 1182, 361–373. [Google Scholar] [CrossRef]
- Liu, J.; Liu, S.; Deng, X.; Rao, J.; Huang, K.; Xu, G.; Wang, X. MicroRNA-582-5p suppresses non-small cell lung cancer cells growth and invasion via downregulating NOTCH1. PLoS ONE 2019, 14, e0217652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, K.-Z.; Chen, W.-W.; Hu, X.-Y.; Jiang, A.-L.; Zhao, J. Clinicopathological and prognostic significance of microRNA-107 in human non small cell lung cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 4545–4551. [Google Scholar] [PubMed]
- Bhattacharyya, N.; Gupta, S.; Sharma, S.; Soni, A.; Bagabir, S.A.; Bhattacharyya, M.; Mukherjee, A.; Almalki, A.H.; Alkhanani, M.F.; Haque, S.; et al. CDK1 and HSP90AA1 Appear as the Novel Regulatory Genes in Non-Small Cell Lung Cancer: A Bioinformatics Approach. J. Pers. Med. 2022, 12, 393. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Liang, B.; Long, F.; Jiang, S.-J. Diagnostic MicroRNA Biomarker Discovery for Non-Small-Cell Lung Cancer Adenocarcinoma by Integrative Bioinformatics Analysis. BioMed Res. Int. 2017, 2017, 2563085. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hao, R.; Zhang, Y.; Deng, H.; Teng, W.; Wang, Z. Construction of circRNA-miRNA-mRNA network and identification of novel potential biomarkers for non-small cell lung cancer. Cancer Cell Int. 2021, 21, 1–7. [Google Scholar] [CrossRef]
- Cai, X.; Lin, L.; Zhang, Q.; Wu, W.; Su, A. Bioinformatics analysis of the circRNA–miRNA–mRNA network for non-small cell lung cancer. J. Int. Med. Res. 2020, 48, 0300060520929167. [Google Scholar] [CrossRef]
- Ding, D.; Zhang, J.; Luo, Z.; Wu, H.; Lin, Z.; Liang, W.; Xue, X. Analysis of the lncRNA–miRNA–mRNA Network Reveals a Potential Regulatory Mechanism of EGFR-TKI Resistance in NSCLC. Front. Genet. 2022, 13, 851391. [Google Scholar] [CrossRef]
- Guo, Q.; Li, D.; Luo, X.; Yuan, Y.; Li, T.; Liu, H.; Wang, X. The Regulatory Network and Potential Role of LINC00973-miRNA-mRNA ceRNA in the Progression of Non-Small-Cell Lung Cancer. Front. Immunol. 2021, 12, 684807. [Google Scholar] [CrossRef]
- Arora, S.; Singh, P.; Ahmad, S.; Ahmad, T.; Dohare, R.; Almatroodi, S.A.; Alrumaihi, F.; Rahmani, A.H.; Syed, M.A. Comprehensive Integrative Analysis Reveals the Association of KLF4 with Macrophage Infiltration and Polarization in Lung Cancer Microenvironment. Cells 2021, 10, 2091. [Google Scholar] [CrossRef]
- Peng, X.; Guan, L.; Gao, B. miRNA-19 promotes non-small-cell lung cancer cell proliferation via inhibiting CBX7 expression. OncoTargets Ther. 2018, 11, 8865–8874. [Google Scholar] [CrossRef] [Green Version]
- Leidinger, P.; Galata, V.; Backes, C.; Stähler, C.; Rheinheimer, S.; Huwer, H.; Meese, E.; Keller, A. Longitudinal study on circulating miRNAs in patients after lung cancer resection. Oncotarget 2015, 6, 16674–16685. [Google Scholar] [CrossRef] [Green Version]
- Shakespear, N.; Ogura, M.; Yamaki, J.; Homma, Y. Astrocyte-Derived Exosomal microRNA miR-200a-3p Prevents MPP+-Induced Apoptotic Cell Death Through Down-Regulation of MKK4. Neurochem. Res. 2020, 45, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Shui, Y.; Yu, X.; Duan, R.; Bao, Q.; Wu, J.; Yuan, H.; Ma, C. miR-130b-3p inhibits cell invasion and migration by targeting the Notch ligand Delta-like 1 in breast carcinoma. Gene 2017, 609, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Kong, C.; Zhu, Y.; Zeng, Y.; Zhang, Z.; Liu, X.; Zhan, B.; Piao, C.; Jiang, Z. miR-130b, an onco-miRNA in bladder cancer, is directly regulated by NF-κB and sustains NF-κB activation by decreasing Cylindromatosis expression. Oncotarget 2016, 7, 48547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Liang, H.; Wang, Y.; Zhang, W.; Zhou, Y.; Chen, S.A.; Yu, M.; Cui, S.; Liu, M.; Wang, N.; et al. miR-96 promotes cell proliferation, migration and invasion by targeting PTPN9 in breast cancer. Sci. Rep. 2016, 6, 37421. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Pu, X.; Wang, Q.; Cao, J.; Xu, F.; Xu, L.; Li, K. miR-96 induces cisplatinchemoresistance in non-small cell lung cancer cells by downregulating SAMD9. Oncol. Lett. 2015, 11, 945–952. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Li, Q.; Li, W.; Zheng, T.; Zhao, S.; Liu, Z. miR-96 downregulates RECK to promote growth and motility of non-small cell lung cancer cells. Mol. Cell. Biochem. 2014, 390, 155–160. [Google Scholar] [CrossRef]
- Yu, S.; Lu, Z.; Liu, C.; Meng, Y.; Ma, Y.; Zhao, W.; Liu, J.; Yu, J.; Chen, J. miRNA-96 Suppresses KRAS and Functions as a Tumor Suppressor Gene in Pancreatic Cancer. Cancer Res. 2010, 70, 6015–6025. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, Y. miR-96 targets SOX6 and promotes proliferation, migration, and invasion of hepatocellular carcinoma. Biochem. Cell Biol. 2018, 96, 365–371. [Google Scholar] [CrossRef]
- Cho, W.C.; Chow, A.S.; Au, J.S. Restoration of tumour suppressor hsa-miR-145 inhibits cancer cell growth in lung adenocarcinoma patients with epidermal growth factor receptor mutation. Eur. J. Cancer 2009, 45, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Miko, E.; Czimmerer, Z.; Csanky, E.; Boros, G.; Buslig, J.; Dezso, B.; Scholtz, B. Differentially expressed microRNAs in small cell lung cancer. Exp. Lung Res. 2009, 35, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Huang, Y.; Zhu, W.; Zhou, S.; Zhou, J.; Zeng, F.; Liu, X.; Zhang, Y.; Yu, J. An integrated analysis of miRNA and mRNA expressions in non-small cell lung cancers. PLoS ONE 2011, 6, e26502. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, P.; Chen, T.; Gao, G.; Chen, X.; Du, Y.; Zhang, R.; Yang, R.; Zhao, W.; Dun, S.F. Expression of microRNA-96 and its potential functions by targeting FOXO3 in non-small cell lung cancer. Tumor Biol. 2015, 36, 685–692. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.W.; Calses, P.; Kemp, C.J.; Taniguchi, T. MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res. 2012, 72, 4037–4046. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Xu, G.; Chang, Z.; Zhu, L.; Yao, J. miR-210 transferred by lung cancer cell-derived exosomes may act as proangiogenic factor in cancer-associated fibroblasts by modulating JAK2/STAT3 pathway. Clin. Sci. 2020, 134, 807–825. [Google Scholar] [CrossRef] [PubMed]
- Katakura, S.; Kobayashi, N.; Hashimoto, H.; Kamimaki, C.; Tanaka, K.; Kubo, S.; Nakashima, K.; Teranishi, S.; Manabe, S.; Watanabe, K.; et al. MicroRNA-200b is a potential biomarker of the expression of PD-L1 in patients with lung cancer. Thorac. Cancer 2020, 11, 2975–2982. [Google Scholar] [CrossRef]
- Lebanony, D.; Benjamin, H.; Gilad, S.; Ezagouri, M.; Dov, A.; Ashkenazi, K.; Gefen, N.; Izraeli, S.; Rechavi, G.; Pass, H.; et al. Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from nonsquamous non–small-cell lung carcinoma. J. Clin. Oncol. 2009, 27, 2030–2037. [Google Scholar] [CrossRef]
- Huang, W.; Jin, Y.; Yuan, Y.; Bai, C.; Wu, Y.; Zhu, H.; Lu, S. Validation and target gene screening of hsa-miR-205 in lung squamous cell carcinoma. Chin. Med. J. 2014, 127, 272–278. [Google Scholar]
- Xiao, K.; Liu, S.; Xiao, Y.; Wang, Y.; Zhu, Z.; Wang, Y.; Tong, D.; Jiang, J. Bioinformatics prediction of differential miRNAs in non-small cell lung cancer. PLoS ONE 2021, 16, e0254854. [Google Scholar] [CrossRef]
- Zhong, M.; Ma, X.; Sun, C.; Chen, L. MicroRNAs reduce tumor growth and contribute to enhance cytotoxicity induced by gefitinib in non-small cell lung cancer. Chem. Interact. 2010, 184, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, H.; Long, L.; Hui, L.; Chen, H.; Wang, X.; Shen, H.; Xu, W. miR-126 enhances the sensitivity of non-small cell lung cancer cells to anticancer agents by targeting vascular endothelial growth factor A. Acta Biochim. Biophys. Sin. 2012, 44, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Qin, W.; Zhang, L.; Hang, J.; Li, B.; Zhang, C.; Wan, J.; Zhou, F.; Shao, K.; Sun, Y.; et al. A 5-microRNA signature for lung squamous cell carcinoma diagnosis and hsa-miR-31 for prognosis. Clin. Cancer Res. 2011, 17, 6802–6811. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.-L.; Wen, Z.-S.; Huang, Y.; Cheng, X.; Wang, G.-Z.; Zhou, Y.-C.; Wang, Z.-Y.; Guo, Y.-Q.; Cao, Y.; Zhou, G.-B. Down-regulation of microRNA-144 in air pollution-related lung cancer. Sci. Rep. 2015, 5, srep14331. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Tian, W.; Zhang, H.; Li, W.; Ji, C.; Wang, Y.; Li, Y. MicroRNA-486-5p Suppresses Lung Cancer via Downregulating mTOR Signaling In Vitro and In Vivo. Front. Oncol. 2021, 11, 1697. [Google Scholar] [CrossRef] [PubMed]
- Raponi, M.; Dossey, L.; Jatkoe, T.; Wu, X.; Chen, G.; Fan, H.; Beer, D.G. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009, 69, 5776–5783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, L.; Todd, N.W.; Yu, L.; Fang, H.; Jiang, F. Early detection of squamous cell lung cancer in sputum by a panel of microRNA markers. Mod. Pathol. 2010, 23, 1157–1164. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Yang, Q.; Wang, X.; Zhou, Y.; Jiang, T.; Ma, Q.; Wang, Y.-J. The role of microRNA in human lung squamous cell carcinoma. Cancer Genet. Cytogenet. 2010, 200, 127–133. [Google Scholar] [CrossRef]
- Yu, L.; Todd, N.W.; Xing, L.; Xie, Y.; Zhang, H.; Liu, Z.; Fang, H.; Zhang, J.; Katz, R.L.; Jiang, F. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int. J. Cancer 2010, 127, 2870–2878. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Shen, H.; Liu, L.; Xu, J.; Xu, J.; Shu, Y. MiR-21 overexpression in human primary squamous cell lung carcinoma is associated with poor patient prognosis. J. Cancer Res. Clin. Oncol. 2010, 137, 557–566. [Google Scholar] [CrossRef]
- Dacic, S.; Kelly, L.; Shuai, Y.; Nikiforova, M.N. miRNA expression profiling of lung adenocarcinomas: Correlation with mutational status. Mod. Pathol. 2010, 23, 1577–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.S.; Jeon, H.-S.; Sun, Z.; Aubry, M.C.; Tang, H.; Park, C.-H.; Rakhshan, F.; Schultz, D.A.; Kolbert, C.P.; Lupu, R.; et al. Increased miR-708 Expression in NSCLC and Its Association with Poor Survival in Lung Adenocarcinoma from Never Smokers. Clin. Cancer Res. 2012, 18, 3658–3667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Brandes, U.; Schoch, D. Centrality as a Predictor of Lethal Proteins: Performance and Robustness. MMB & DFT 2014, 2014, 11. [Google Scholar]
- Hake, L.E.; Richter, J.D. CPEB is a specificity factor that mediates cytoplasmic polyadenylation during Xenopus oocyte maturation. Cell 1994, 79, 617–627. [Google Scholar] [CrossRef]
- Fang, Y.; Zhong, Q.; Wang, Y.; Gu, C.; Liu, S.; Li, A.; Yan, Q. CPEB3 functions as a tumor suppressor in colorectal cancer via JAK/STAT signaling. Aging 2020, 12, 21404–21422. [Google Scholar] [CrossRef]
- Hansen, C.N.; Ketabi, Z.; Rosenstierne, M.W.; Palle, C.; Boesen, H.C.; Norrild, B. Expression of CPEB, GAPDH and U6snRNA in cervical and ovarian tissue during cancer development. APMIS 2009, 117, 53–59. [Google Scholar] [CrossRef]
- D’Ambrogio, A.; Nagaoka, K.; Richter, J.D. Translational control of cell growth and malignancy by the CPEBs. Nat. Cancer 2013, 13, 283–290. [Google Scholar] [CrossRef]
- Sheng, H.; Li, X.; Xu, Y. Knockdown of FOXP1 promotes the development of lung adenocarcinoma. Cancer Biol. Ther. 2018, 20, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.J.; Seo, E.J.; Kim, D.K.; Lee, S.I.; Kwon, Y.W.; Jang, I.H.; Kim, K.-H.; Suh, D.-S.; Kim, J.H. FOXP1 functions as an oncogene in promoting cancer stem cell-like characteristics in ovarian cancer cells. Oncotarget 2015, 7, 3506–3519. [Google Scholar] [CrossRef] [Green Version]
- Koon, H.B.; Ippolito, G.C.; Banham, A.; Tucker, P.W. FOXP1: A potential therapeutic target in cancer. Expert Opin. Ther. Targets 2007, 11, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liao, Y.; Tang, M.; Wu, T.; Tan, D.; Zhang, S.; Wang, H. Trps1 is associated with the multidrug resistance of lung cancer cell by regulating MGMT gene expression. Cancer Med. 2018, 7, 1921–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giedion, A.; Burdea, M.; Fruchter, Z.; Meloni, T.; Trosc, V. Autosomal-dominant transmission of the tricho-rhino-phalangeal syndrome. Report of 4 unrelated families, review of 60 cases. Helv. Paediatr. Acta 1973, 28, 249–259. [Google Scholar]
- Momeni, P.; Glöckner, G.; Schmidt, O.; Von Holtum, D.; Albrecht, B.; Gillessen-Kaesbach, G.; Hennekam, R.; Meinecke, P.; Zabel, B.; Rosenthal, A.; et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat. Genet. 2000, 24, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.T.G.; Jhamai, M.; Van Weerden, W.M.; Jenster, G.; Brinkmann, A.O. The TRPS1 transcription factor: Androgenic regulation in prostate cancer and high expression in breast cancer. Endocr. -Relat. Cancer 2004, 11, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. miR-221/222 targeting of trichorhinophalangeal 1 (TRPS1) promotes epithelial-to-mesenchymal transition in breast cancer. Sci. Signal. 2011, 4, pt5. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jia, M.; Wu, X.; Cui, J.; Pan, A.; Li, L. Overexpression of Trps1 contributes to tumor angiogenesis and poor prognosis of human osteosarcoma. Diagn. Pathol. 2015, 10, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantauzzo, K.A.; Kurban, M.; Levy, B.; Christiano, A.M. Trps1 and Its Target Gene Sox9 Regulate Epithelial Proliferation in the Developing Hair Follicle and Are Associated with Hypertrichosis. PLoS Genet. 2012, 8, e1003002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gai, Z.; Zhou, G.; Gui, T.; Itoh, S.; Oikawa, K.; Uetani, K.; Muragaki, Y. Trps1 Haploinsufficiency Promotes Renal Fibrosis by Increasing Arkadia Expression. J. Am. Soc. Nephrol. 2010, 21, 1468–1476. [Google Scholar] [CrossRef] [Green Version]
- Cy, L. Expression of T cell factor-4 in non-small-cell lung cancer. Chin. Med. J. 2005, 118, 136–140. [Google Scholar]
- Zhang, X.-W.; Chen, S.-Y.; Xue, D.-W.; Xu, H.-H.; Yang, L.-H.; Xu, H.-T.; Wang, E.-H. Expression of Nemo-like kinase was increased and negatively correlated with the expression of TCF4 in lung cancers. Int. J. Clin. Exp. Pathol. 2015, 8, 15086–15092. [Google Scholar]
- Angus-Hill, M.L.; Elbert, K.M.; Hidalgo, J.; Capecchi, M.R. T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 4914–4919. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Cai, J.; Xie, S.; Zhao, C.; Wang, Y.; Cao, D.; Li, G. T Cell Factor 4 Is Involved in Papillary Thyroid Carcinoma via Regulating Long Non-Coding RNA HCP5. Technol. Cancer Res. Treat. 2020, 19, 1533033820983290. [Google Scholar] [CrossRef]
- Perissi, V.; Scafoglio, C.; Zhang, J.; Ohgi, K.A.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. TBL1 and TBLR1 Phosphorylation on Regulated Gene Promoters Overcomes Dual CtBP and NCoR/SMRT Transcriptional Repression Checkpoints. Mol. Cell 2008, 29, 755–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Liu, C.; Yu, Y.; Geng, J.; Meng, Q.; Xu, S.; Zhou, F.; Chen, Y.; Jin, S.; Shen, J.; et al. TBL1XR1 is involved in c-Met-mediated tumorigenesis of human nonsmall cell lung cancer. Cancer Gene Ther. 2019, 27, 136–146. [Google Scholar] [CrossRef]
- Pasmant, E.; Gilbert-Dussardier, B.; Petit, A.; de Laval, B.; Luscan, A.; Gruber, A.; Lapillonne, H.; Deswarte, C.; Goussard, P.; Laurendeau, I.; et al. SPRED1, a RAS MAPK pathway inhibitor that causes Legius syndrome, is a tumour suppressor downregulated in paediatric acute myeloblasticleukaemia. Oncogene 2014, 34, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Ladd, A.N. The importance of CELF control: Molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. WileyInterdiscip. Rev. RNA 2012, 3, 104–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piqué, L.; Martinez de Paz, A.; Piñeyro, D.; Martínez-Cardús, A.; Castro de Moura, M.; Llinàs-Arias, P.; Setien, F.; Gomez-Miragaya, J.; Gonzalez-Suarez, E.; Sigurdsson, S.; et al. Epigenetic inactivation of the splicing RNA-binding protein CELF2 in human breast cancer. Oncogene 2019, 38, 7106–7112. [Google Scholar] [CrossRef]
- Chang, Y.-S.; Tu, S.-J.; Chiang, H.-S.; Yen, J.-C.; Lee, Y.-T.; Fang, H.-Y.; Chang, J.-G. Genome-Wide Analysis of Prognostic Alternative Splicing Signature and Splicing Factors in Lung Adenocarcinoma. Genes 2020, 11, 1300. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.T.; Fan, S.; Lu, B.; Yin, S.; Yang, S.; Nie, W.; Wang, M.; Zhou, L.; Li, T.; Li, X.; et al. CELF2 suppresses non-small cell lung carcinoma growth by inhibiting the PREX2-PTEN interaction. Carcinogenesis 2020, 41, 377–389. [Google Scholar] [CrossRef]
- Łukasik, P.; Załuski, M.; Gutowska, I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development–Review. Int. J. Mol. Sci. 2021, 22, 2935. [Google Scholar] [CrossRef]
- Dannappel, M.V.; Sooraj, D.; Loh, J.J.; Firestein, R. Molecular and in vivo Functions of the CDK8 and CDK19 Kinase Modules. Front. Cell Dev. Biol. 2019, 6, 171. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 Homolog at 3q27–29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Bergholz, J.; Xiao, Z.-X. Role of p63 in Development, Tumorigenesis and Cancer Progression. Cancer Microenviron. 2012, 5, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Lisztwan, J.; Imbert, G.; Wirbelauer, C.; Gstaiger, M.; Krek, W. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 1999, 13, 1822–1833. [Google Scholar] [CrossRef]
- Tan, R.J.; Zhou, D.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling and kidney fibrosis. Kidney Int. Suppl. 2014, 4, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.-M.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 2017, 7, 1389–1406. [Google Scholar]
- Kudo, Y.; Tsunematsu, T.; Takata, T. Oncogenic role of RUNX3 in head and neck cancer. J. Cell. Biochem. 2011, 112, 387–393. [Google Scholar] [CrossRef]
- Chen, F.; Liu, X.; Bai, J.; Pei, D.; Zheng, J. The emerging role of RUNX3 in cancer metastasis (Review). Oncol. Rep. 2015, 35, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Gustafsson, J. The different roles of ER subtypes in cancer biology and therapy. Nat. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef]
- Wang, B.; Guo, H.; Yu, H.; Chen, Y.; Xu, H.; Zhao, G. The Role of the Transcription Factor EGR1 in Cancer. Front. Oncol. 2021, 11, 642547. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Liu, X.S.; Carroll, J.; Jänne, O.A.; Keeton, E.K.; Chinnaiyan, A.M.; Pienta, K.; Brown, M. A Hierarchical Network of Transcription Factors Governs Androgen Receptor-Dependent Prostate Cancer Growth. Mol. Cell 2007, 27, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Ishrat, R.; Tazyeen, S.; Alam, A.; Farooqui, A.; Ali, R.; Imam, N.; Tamkeen, N.; Ali, S.; Malik, M.Z.; et al. In silico integrative approach revealed key microRNAs and associated target genes in cardiorenal syndrome. Bioinform. Biol. Insights 2021, 15, 11779322211027396. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Tazyeen, S.; Ali, R.; Alam, A.; Imam, N.; Malik, M.Z.; Ali, S.; Ishrat, R. Network centrality approaches used to uncover and classify most influential nodes with their related miRNAs in cardiovascular diseases. Gene Rep. 2022, 27, 101555. [Google Scholar] [CrossRef]
- Ahmed, M.; Singh, P.; Sultan, A.; Dohare, R.; Tazyeen, S.; Alam, A.; Ali, R.; Farooqui, A.; Imam, N.; Tamkeen, N.; et al. Unravelling the role of hub genes associated with cardio renal syndrome through an integrated bioinformatics approach. Gene Rep. 2021, 25, 101382. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Series | TS | N | D | UR | DR | GPL | C | Y |
|---|---|---|---|---|---|---|---|---|
| GSE25508 | 60 | 26 | 34 | 39 | 52 | 7731 | Finland | 2011 |
| GSE19945 | 63 | 8 | 55 | 14 | 31 | 9948 | Japan | 2013 |
| GSE53882 | 548 | 397 | 151 | 29 | 7 | 18130 | China | 2017 |
| Adjusted p-Value | p-Value | Log FC | miRNA | OG | OG vs. CTD |
|---|---|---|---|---|---|
| 0.000488 | 3.6 × 10−7 | 3.44103 | MiR-30a | 1076 | 1050 |
| 0.002008 | 4.63 × 10−6 | 4.13116 | MiR-145 | 154 | 149 |
| 0.002008 | 4.59 × 10−6 | 1.95769 | MiR-140-3p | 387 | 370 |
| 0.002008 | 6.86 × 10−6 | 2.22649 | MiR-572 | 122 | 118 |
| 0.002008 | 7.9 × 10−6 | 1.68913 | MiR-144 | 144 | 137 |
| 0.004767 | 3.24 × 10−5 | 2.08135 | MiR-126 | 11 | 10 |
| 0.008815 | 1.93 × 10−4 | 2.66117 | MiR-486-5p | 99 | 95 |
| 0.014676 | 1.13 × 10−3 | −1.71609 | MiR-210 | 26 | 26 |
| 0.014796 | 1.17 × 10−3 | −1.88279 | MiR-130b | 592 | 586 |
| 0.018155 | 2.38 × 10−3 | −1.65166 | MiR-96 | 290 | 285 |
| 0.004867 | 4.90 × 10−6 | −0.71609 | MiR-200b | 573 | 560 |
| 0.006767 | 7.90 × 10−6 | −0.65166 | MiR-205 | 832 | 800 |
| Term | p-Value | Adjusted p-Value | Key Genes |
|---|---|---|---|
| TP63 human | 0.006731046 | 0.029080631 | TCF4 |
| VHL human | 0.008965773 | 0.029080631 | TCF4 |
| LEF1 human | 0.012977027 | 0.029080631 | TCF4 |
| RUNX3 human | 0.013421829 | 0.029080631 | TCF4 |
| ESR1 human | 0.033691182 | 0.044511811 | FOXP1 |
| EGR1 human | 0.038917584 | 0.044511811 | TCF4 |
| AR human | 0.041087826 | 0.044511811 | TRPS1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shafat, Z.; Ahmed, M.M.; Almajhdi, F.N.; Hussain, T.; Parveen, S.; Ahmed, A. Identification of the Key miRNAs and Genes Associated with the Regulation of Non-Small Cell Lung Cancer: A Network-Based Approach. Genes 2022, 13, 1174. https://doi.org/10.3390/genes13071174
Shafat Z, Ahmed MM, Almajhdi FN, Hussain T, Parveen S, Ahmed A. Identification of the Key miRNAs and Genes Associated with the Regulation of Non-Small Cell Lung Cancer: A Network-Based Approach. Genes. 2022; 13(7):1174. https://doi.org/10.3390/genes13071174
Chicago/Turabian StyleShafat, Zoya, Mohd Murshad Ahmed, Fahad N. Almajhdi, Tajamul Hussain, Shama Parveen, and Anwar Ahmed. 2022. "Identification of the Key miRNAs and Genes Associated with the Regulation of Non-Small Cell Lung Cancer: A Network-Based Approach" Genes 13, no. 7: 1174. https://doi.org/10.3390/genes13071174
APA StyleShafat, Z., Ahmed, M. M., Almajhdi, F. N., Hussain, T., Parveen, S., & Ahmed, A. (2022). Identification of the Key miRNAs and Genes Associated with the Regulation of Non-Small Cell Lung Cancer: A Network-Based Approach. Genes, 13(7), 1174. https://doi.org/10.3390/genes13071174