
microRNA Expression Profile of Purified Alveolar Epithelial Type II Cells

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Preparation of Single-Cell Suspensions and Cell Sorting
2.3. Flow Cytometry and Immunofluorescence Staining for Purity Assessment
2.4. Isolation of ATII Cells (Panning)
2.5. Papanicolaou Staining
2.6. TGF-β1 Stimulation of A549 Cells
2.7. RNA Isolation
2.8. Reverse Transcription and Quantitative PCR of mRNAs
2.9. Reverse Transcription and Quantitative PCR of miRNAs
2.10. Analysis of TaqMan® Real-Time PCR miRNA Array Data
3. Results
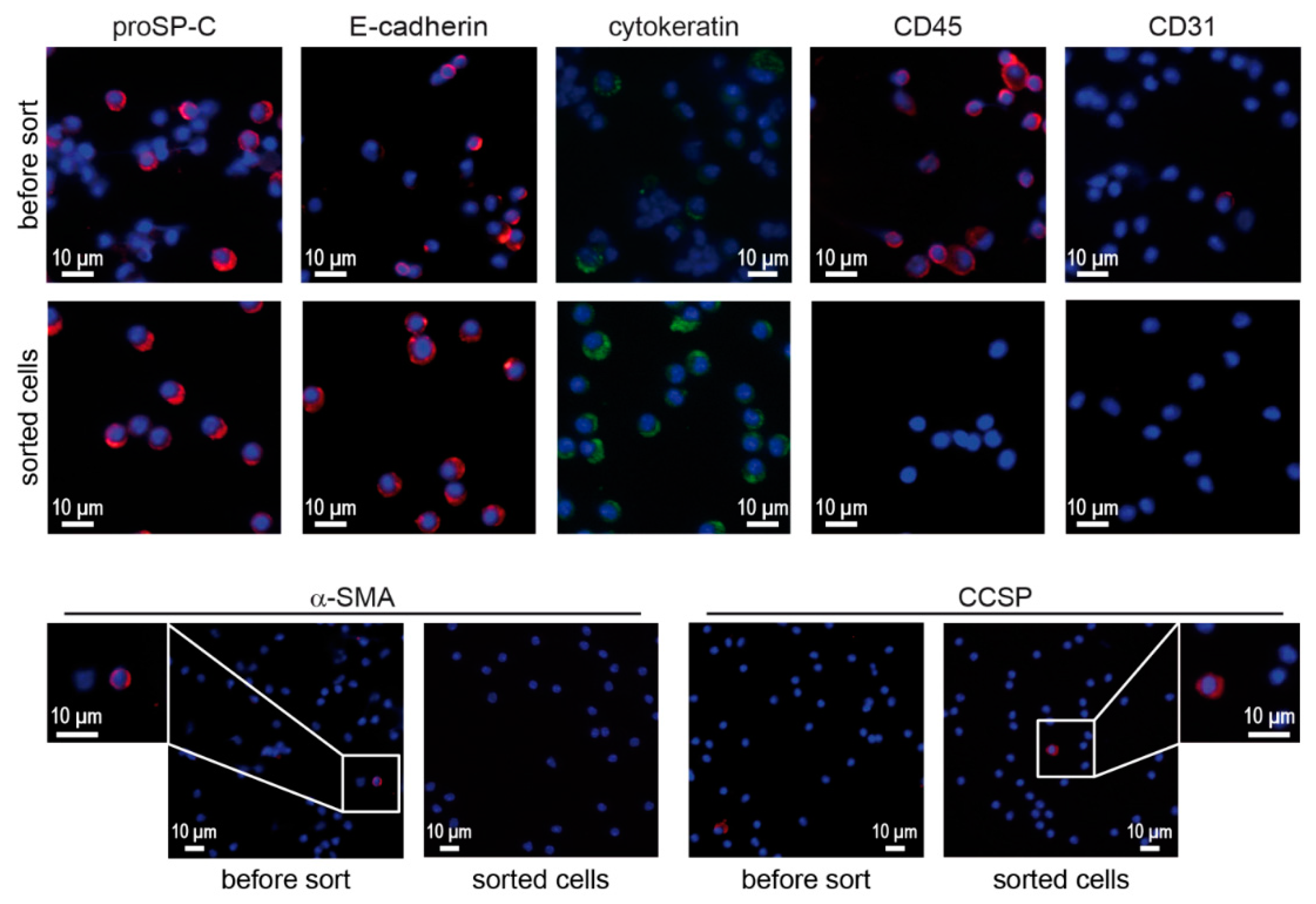
3.1. Isolation by Sorting and Assessment of Purity of Primary Murine ATII Cells
3.2. Confirmation of Epithelial and ATII Identity of the Sorted Cell Population
3.3. Viability, Purity and Phenotypes of ATII Cells Isolated by Sorting and Panning
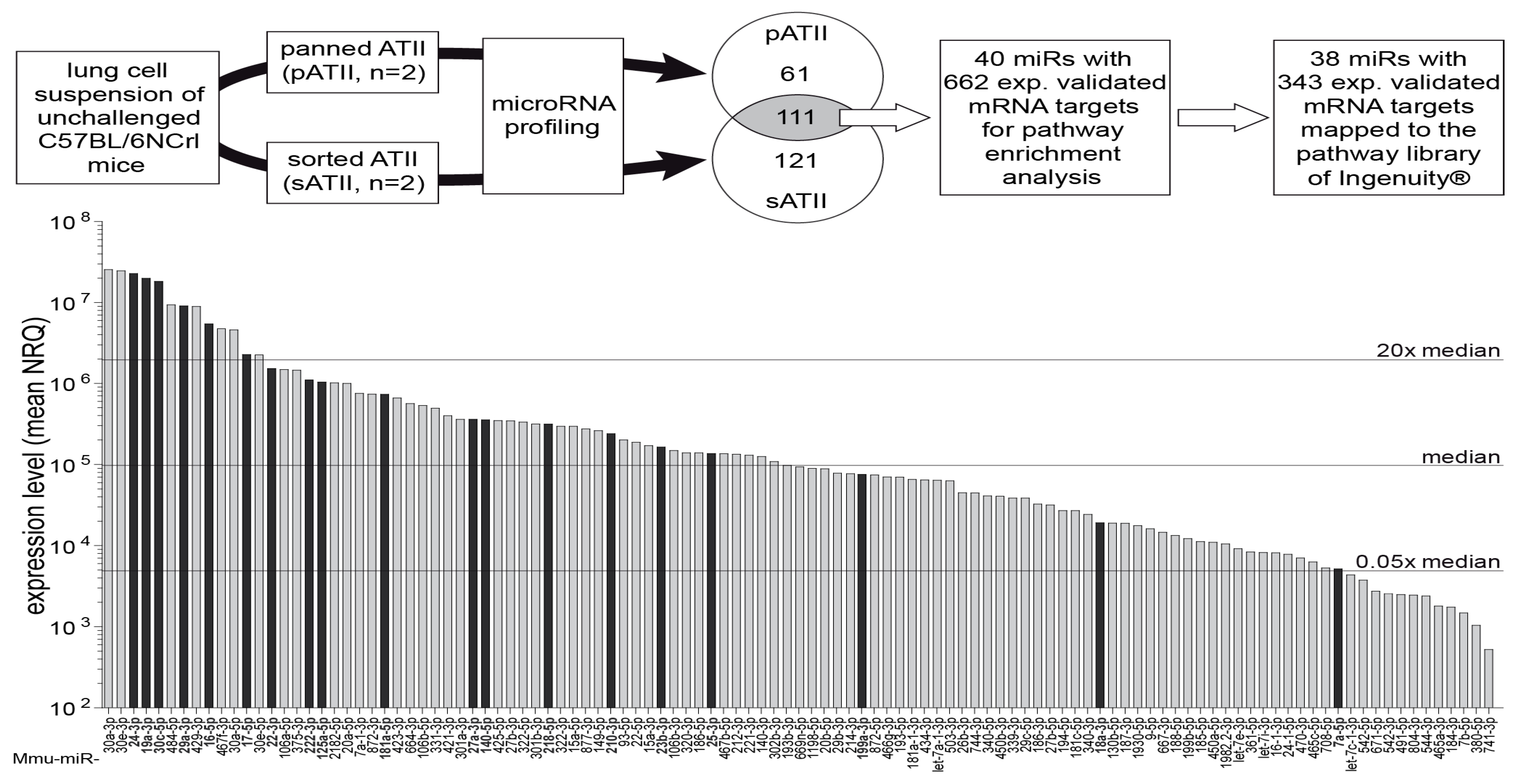
3.4. MiRNA Expression Profiling of ATII Cells and Pathway Enrichment Analysis of Downstream mRNA Targets
3.5. Key Upstream Regulators of Target mRNAs
3.6. mRNA Targets Located within the TGF-β Signaling Pathway
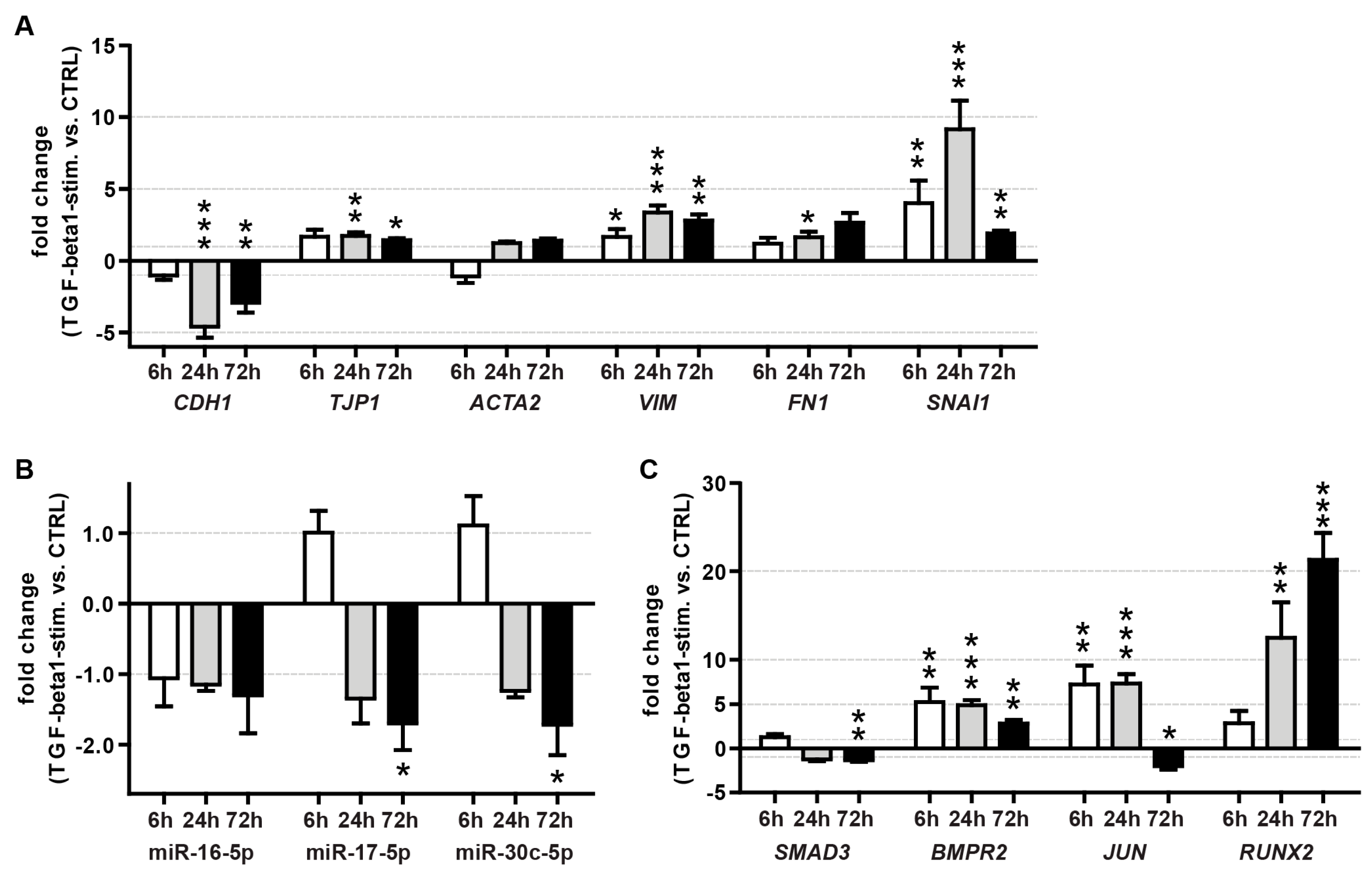
3.6.1. Effect of TGF-β1 Treatment on miRNA Expression in A549 Cells
3.6.2. Effect of TGF-β1 Treatment on Target mRNA Expression in A549 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Berezikov, E. Evolution of microRNA diversity and regulation in animals. Nat. Rev. Genet. 2011, 12, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; Abdellatif, M. MicroRNAs in development and disease. Physiol. Rev. 2011, 91, 827–887. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Rahman, M.; Nana-Sinkam, S.P. MicroRNAs in respiratory disease. A clinician’s overview. Ann. Am. Thorac. Soc. 2014, 11, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.E.; Moschos, S.A.; Perry, M.M.; Barnes, P.J.; Lindsay, M.A. Maternally imprinted microRNAs are differentially expressed during mouse and human lung development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.E.; Perry, M.M.; Moschos, S.A.; Lindsay, M.A. microRNA expression in the aging mouse lung. BMC Genom. 2007, 8, 172. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Frank, D.B.; Morley, M.P.; Zhou, S.; Wang, X.; Lu, M.M.; Lazar, M.A.; Morrisey, E.E. HDAC3-Dependent Epigenetic Pathway Controls Lung Alveolar Epithelial Cell Remodeling and Spreading via miR-17-92 and TGF-beta Signaling Regulation. Dev. Cell 2016, 36, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Mathis, C.; Poussin, C.; Weisensee, D.; Gebel, S.; Hengstermann, A.; Sewer, A.; Belcastro, V.; Xiang, Y.; Ansari, S.; Wagner, S.; et al. Human bronchial epithelial cells exposed in vitro to cigarette smoke at the air-liquid interface resemble bronchial epithelium from human smokers. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L489–L503. [Google Scholar] [CrossRef]
- Huang, Y.; Crawford, M.; Higuita-Castro, N.; Nana-Sinkam, P.; Ghadiali, S.N. miR-146a regulates mechanotransduction and pressure-induced inflammation in small airway epithelium. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 3351–3364. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, X.R.; Wei, L.H.; Chung, A.C.; Yu, C.M.; Lan, H.Y. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 974–985. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, J.; Chen, J.; Feng, T.; Guo, Q. MiR-29 mediates TGFbeta 1-induced extracellular matrix synthesis through activation of Wnt/beta -catenin pathway in human pulmonary fibroblasts. Technol. Health Care Off. J. Eur. Soc. Eng. Med. 2015, 23 (Suppl. 1), S119–S125. [Google Scholar]
- Yang, S.; Banerjee, S.; de Freitas, A.; Sanders, Y.Y.; Ding, Q.; Matalon, S.; Thannickal, V.J.; Abraham, E.; Liu, G. Participation of miR-200 in pulmonary fibrosis. Am. J. Pathol. 2012, 180, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Liang, J.; Geng, Y.; Liu, N.; Kurkciyan, A.; Kulur, V.; Leng, D.; Deng, N.; Liu, Z.; Song, J.; et al. MicroRNA-29c Prevents Pulmonary Fibrosis by Regulating Epithelial Cell Renewal and Apoptosis. Am. J. Respir. Cell Mol. Biol. 2017, 57, 721–732. [Google Scholar] [CrossRef]
- Milosevic, J.; Pandit, K.; Magister, M.; Rabinovich, E.; Ellwanger, D.C.; Yu, G.; Vuga, L.J.; Weksler, B.; Benos, P.V.; Gibson, K.F.; et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Kho, A.T.; McGeachie, M.J.; Moore, K.G.; Sylvia, J.M.; Weiss, S.T.; Tantisira, K.G. Circulating microRNAs and prediction of asthma exacerbation in childhood asthma. Respir. Res. 2018, 19, 128. [Google Scholar] [CrossRef] [Green Version]
- Ezzie, M.E.; Crawford, M.; Cho, J.H.; Orellana, R.; Zhang, S.; Gelinas, R.; Batte, K.; Yu, L.; Nuovo, G.; Galas, D.; et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax 2012, 67, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Conickx, G.; Mestdagh, P.; Avila Cobos, F.; Verhamme, F.M.; Maes, T.; Vanaudenaerde, B.M.; Seys, L.J.; Lahousse, L.; Kim, R.Y.; Hsu, A.C.; et al. MicroRNA Profiling Reveals a Role for MicroRNA-218-5p in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 43–56. [Google Scholar] [CrossRef]
- Quan, Y.; Wang, Z.; Gong, L.; Peng, X.; Richard, M.A.; Zhang, J.; Fornage, M.; Alcorn, J.L.; Wang, D. Exosome miR-371b-5p promotes proliferation of lung alveolar progenitor type II cells by using PTEN to orchestrate the PI3K/Akt signaling. Stem Cell Res. Ther. 2017, 8, 138. [Google Scholar] [CrossRef]
- Mason, R.J. Biology of alveolar type II cells. Respirology 2006, 11, S12–S15. [Google Scholar] [CrossRef]
- Weibel, E.R. On the tricks alveolar epithelial cells play to make a good lung. Am. J. Respir. Crit. Care Med. 2015, 191, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Kapanci, Y.; Weibel, E.R.; Kaplan, H.P.; Robinson, F.R. Pathogenesis and reversibility of the pulmonary lesions of oxygen toxicity in monkeys. II. Ultrastructural and morphometric studies. Lab. Investig. A J. Tech. Methods Pathol. 1969, 20, 101–118. [Google Scholar]
- Hirai, K.I.; Witschi, H.; Cote, M.G. Electron microscopy of butylated hydroxytoluene-induced lung damage in mice. Exp. Mol. Pathol. 1977, 27, 295–308. [Google Scholar] [CrossRef]
- Zemans, R.L.; Briones, N.; Campbell, M.; McClendon, J.; Young, S.K.; Suzuki, T.; Yang, I.V.; De Langhe, S.; Reynolds, S.D.; Mason, R.J.; et al. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 15990–15995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanjore, H.; Degryse, A.L.; Crossno, P.F.; Xu, X.C.; McConaha, M.E.; Jones, B.R.; Polosukhin, V.V.; Bryant, A.J.; Cheng, D.S.; Newcomb, D.C.; et al. beta-catenin in the alveolar epithelium protects from lung fibrosis after intratracheal bleomycin. Am. J. Respir. Crit. Care Med. 2013, 187, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Aso, Y.; Yoneda, K.; Kikkawa, Y. Morphologic and biochemical study of pulmonary changes induced by bleomycin in mice. Lab. Investig. A J. Tech. Methods Pathol. 1976, 35, 558–568. [Google Scholar]
- Zhang, Y.; Goss, A.M.; Cohen, E.D.; Kadzik, R.; Lepore, J.J.; Muthukumaraswamy, K.; Yang, J.; DeMayo, F.J.; Whitsett, J.A.; Parmacek, M.S.; et al. A Gata6-Wnt pathway required for epithelial stem cell development and airway regeneration. Nat. Genet. 2008, 40, 862–870. [Google Scholar] [CrossRef] [Green Version]
- Aoshiba, K.; Yokohori, N.; Nagai, A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am. J. Respir. Cell Mol. Biol. 2003, 28, 555–562. [Google Scholar] [CrossRef]
- Evans, M.J.; Cabral, L.J.; Stephens, R.J.; Freeman, G. Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp. Mol. Pathol. 1975, 22, 142–150. [Google Scholar] [CrossRef]
- Bowden, D.H.; Davies, E.; Wyatt, J.P. Cytodynamics of pulmonary alveolar cells in the mouse. Arch. Pathol. 1968, 86, 667–670. [Google Scholar] [PubMed]
- Kauffman, S.L. Cell proliferation in the mammalian lung. Int. Rev. Exp. Pathol. 1980, 22, 131–191. [Google Scholar]
- Adamson, I.Y.; Bowden, D.H. The type 2 cell as progenitor of alveolar epithelial regeneration. A cytodynamic study in mice after exposure to oxygen. Lab. Investig. A J. Tech. Methods Pathol. 1974, 30, 35–42. [Google Scholar]
- Tesfaigzi, J.; Wood, M.B.; Johnson, N.F.; Nikula, K.J. Apoptosis is a pathway responsible for the resolution of endotoxin-induced alveolar type II cell hyperplasia in the rat. Int. J. Exp. Pathol. 1998, 79, 303–311. [Google Scholar] [CrossRef]
- Bardales, R.H.; Xie, S.S.; Schaefer, R.F.; Hsu, S.M. Apoptosis is a major pathway responsible for the resolution of type II pneumocytes in acute lung injury. Am. J. Pathol. 1996, 149, 845–852. [Google Scholar]
- Motz, G.T.; Eppert, B.L.; Wesselkamper, S.C.; Flury, J.L.; Borchers, M.T. Chronic cigarette smoke exposure generates pathogenic T cells capable of driving COPD-like disease in Rag2−/− mice. Am. J. Respir. Crit. Care Med. 2010, 181, 1223–1233. [Google Scholar] [CrossRef] [Green Version]
- Yokohori, N.; Aoshiba, K.; Nagai, A.; Respiratory Failure Research Group in, J. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest 2004, 125, 626–632. [Google Scholar] [CrossRef]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef]
- Bhandary, Y.P.; Shetty, S.K.; Marudamuthu, A.S.; Gyetko, M.R.; Idell, S.; Gharaee-Kermani, M.; Shetty, R.S.; Starcher, B.C.; Shetty, S. Regulation of alveolar epithelial cell apoptosis and pulmonary fibrosis by coordinate expression of components of the fibrinolytic system. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L463–L473. [Google Scholar] [CrossRef] [Green Version]
- Crapo, J.D.; Barry, B.E.; Gehr, P.; Bachofen, M.; Weibel, E.R. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982, 126, 332–337. [Google Scholar]
- Ridsdale, R.; Post, M. Surfactant lipid synthesis and lamellar body formation in glycogen-laden type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L743–L751. [Google Scholar] [CrossRef]
- Evans, M.J.; Hackney, J.D. Cell proliferation in lungs of mice exposed to elevated concentrations of oxygen. Aerosp. Med. 1972, 43, 620–622. [Google Scholar]
- Liu, Y.; Sadikot, R.T.; Adami, G.R.; Kalinichenko, V.V.; Pendyala, S.; Natarajan, V.; Zhao, Y.Y.; Malik, A.B. FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J. Exp. Med. 2011, 208, 1473–1484. [Google Scholar] [CrossRef] [Green Version]
- Jansing, N.L.; McClendon, J.; Henson, P.M.; Tuder, R.M.; Hyde, D.M.; Zemans, R.L. Unbiased Quantitation of Alveolar Type II to Alveolar Type I Cell Transdifferentiation during Repair after Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2017, 57, 519–526. [Google Scholar] [CrossRef]
- Andreeva, A.V.; Kutuzov, M.A.; Voyno-Yasenetskaya, T.A. Regulation of surfactant secretion in alveolar type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L259–L271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelps, D.S.; Floros, J. Localization of pulmonary surfactant proteins using immunohistochemistry and tissue in situ hybridization. Exp. Lung Res. 1991, 17, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Ariki, S.; Kojima, T.; Gasa, S.; Saito, A.; Nishitani, C.; Takahashi, M.; Shimizu, T.; Kurimura, Y.; Sawada, N.; Fujii, N.; et al. Pulmonary collectins play distinct roles in host defense against Mycobacterium avium. J. Immunol. 2011, 187, 2586–2594. [Google Scholar] [CrossRef] [Green Version]
- Carreto-Binaghi, L.E.; Aliouat, M.E.; Taylor, M.L. Surfactant proteins, SP-A and SP-D, in respiratory fungal infections: Their role in the inflammatory response. Respir. Res. 2016, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, M.; Scoville, E.A.; Grant, S.; Korfhagen, T.; Brondyk, W.; Scheule, R.K.; Whitsett, J.A. Surfactant protein-D and surfactant inhibit endotoxin-induced pulmonary inflammation. Chest 2007, 132, 1447–1454. [Google Scholar] [CrossRef]
- Nogee, L.M.; Garnier, G.; Dietz, H.C.; Singer, L.; Murphy, A.M.; deMello, D.E.; Colten, H.R. A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J. Clin. Investig. 1994, 93, 1860–1863. [Google Scholar] [CrossRef]
- Nogee, L.M.; de Mello, D.E.; Dehner, L.P.; Colten, H.R. Brief report: Deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N. Engl. J. Med. 1993, 328, 406–410. [Google Scholar] [CrossRef]
- Glasser, S.W.; Detmer, E.A.; Ikegami, M.; Na, C.L.; Stahlman, M.T.; Whitsett, J.A. Pneumonitis and emphysema in sp-C gene targeted mice. J. Biol. Chem. 2003, 278, 14291–14298. [Google Scholar] [CrossRef] [Green Version]
- Glasser, S.W.; Witt, T.L.; Senft, A.P.; Baatz, J.E.; Folger, D.; Maxfield, M.D.; Akinbi, H.T.; Newton, D.A.; Prows, D.R.; Korfhagen, T.R. Surfactant protein C-deficient mice are susceptible to respiratory syncytial virus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L64–L72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasser, S.W.; Senft, A.P.; Maxfield, M.D.; Ruetschilling, T.L.; Baatz, J.E.; Page, K.; Korfhagen, T.R. Genetic replacement of surfactant protein-C reduces respiratory syncytial virus induced lung injury. Respir. Res. 2013, 14, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitra, M.; Cano, C.A.; Garcia, C.K. Mutant surfactant A2 proteins associated with familial pulmonary fibrosis and lung cancer induce TGF-beta1 secretion. Proc. Natl. Acad. Sci. USA 2012, 109, 21064–21069. [Google Scholar] [CrossRef] [Green Version]
- Konigshoff, M.; Kramer, M.; Balsara, N.; Wilhelm, J.; Amarie, O.V.; Jahn, A.; Rose, F.; Fink, L.; Seeger, W.; Schaefer, L.; et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Investig. 2009, 119, 772–787. [Google Scholar] [CrossRef] [Green Version]
- Corti, M.; Brody, A.R.; Harrison, J.H. Isolation and primary culture of murine alveolar type II cells. Am. J. Respir. Cell Mol. Biol. 1996, 14, 309–315. [Google Scholar] [CrossRef]
- Dobbs, L.G. Isolation and culture of alveolar type II cells. Am. J. Physiol. 1990, 258 Pt 1, L134–L147. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001, 29, e45. [Google Scholar] [CrossRef]
- Iglewicz, B.; Hoaglin, D. How to detect and handle outliers. In The ASQC Basic References in Quality Control: Statistical Techniques; Mykytka, E.F., Ed.; Asq Press: Milwaukee, WI, USA, 1993; Volume 16. [Google Scholar]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, A.C.; Milne, D.S.; Wilkes, J.; Dark, J.H.; Tetley, T.D.; Kirby, J.A. Constitutive expression of MHC and adhesion molecules by alveolar epithelial cells (type II pneumocytes) isolated from human lung and comparison with immunocytochemical findings. J. Cell Sci. 1994, 107 Pt 2, 443–449. [Google Scholar] [CrossRef]
- Marsh, L.M.; Cakarova, L.; Kwapiszewska, G.; von Wulffen, W.; Herold, S.; Seeger, W.; Lohmeyer, J. Surface expression of CD74 by type II alveolar epithelial cells: A potential mechanism for macrophage migration inhibitory factor-induced epithelial repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L442–L452. [Google Scholar] [CrossRef] [Green Version]
- Gereke, M.; Jung, S.; Buer, J.; Bruder, D. Alveolar type II epithelial cells present antigen to CD4(+) T cells and induce Foxp3(+) regulatory T cells. Am. J. Respir. Crit. Care Med. 2009, 179, 344–355. [Google Scholar] [CrossRef]
- Chacko, A.M.; Nayak, M.; Greineder, C.F.; Delisser, H.M.; Muzykantov, V.R. Collaborative enhancement of antibody binding to distinct PECAM-1 epitopes modulates endothelial targeting. PLoS ONE 2012, 7, e34958. [Google Scholar] [CrossRef] [Green Version]
- Ingenuity® Systems, Summer Release 2012. Available online: www.ingenuity.com (accessed on 11 June 2022).
- Choi, J.E.; Lee, S.S.; Sunde, D.A.; Huizar, I.; Haugk, K.L.; Thannickal, V.J.; Vittal, R.; Plymate, S.R.; Schnapp, L.M. Insulin-like growth factor-I receptor blockade improves outcome in mouse model of lung injury. Am. J. Respir. Crit. Care Med. 2009, 179, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Kavvadas, P.; Kypreou, K.P.; Protopapadakis, E.; Prodromidi, E.; Sideras, P.; Charonis, A.S. Integrin-linked kinase (ILK) in pulmonary fibrosis. Virchows Arch. Int. J. Pathol. 2010, 457, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Le Cras, T.D.; Korfhagen, T.R.; Davidson, C.; Schmidt, S.; Fenchel, M.; Ikegami, M.; Whitsett, J.A.; Hardie, W.D. Inhibition of PI3K by PX-866 prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am. J. Pathol. 2010, 176, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, N.; Kuwano, K.; Maeyama, T.; Hagimoto, N.; Yoshimi, M.; Hamada, N.; Yamada, M.; Nakanishi, Y. The p53-Mdm2 association in epithelial cells in idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. J. Clin. Pathol. 2005, 58, 583–589. [Google Scholar] [CrossRef]
- Son, G.; Hines, I.N.; Lindquist, J.; Schrum, L.W.; Rippe, R.A. Inhibition of phosphatidylinositol 3-kinase signaling in hepatic stellate cells blocks the progression of hepatic fibrosis. Hepatology 2009, 50, 1512–1523. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Khalil, W.; Kahm, J.; Jessurun, J.; Kleidon, J.; Henke, C.A. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am. J. Pathol. 2010, 176, 2626–2637. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.L.; Cottrell, E.; Hoban, P.R.; Spiteri, M.A. RhoA signaling modulates cyclin D1 expression in human lung fibroblasts; implications for idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.P.; Gregory, L.G.; Causton, B.; Campbell, G.A.; Lloyd, C.M. Activin A and TGF-beta promote T(H)9 cell-mediated pulmonary allergic pathology. J. Allergy Clin. Immunol. 2012, 129, 1000–1010.e1003. [Google Scholar] [CrossRef] [Green Version]
- Konigshoff, M.; Kneidinger, N.; Eickelberg, O. TGF-beta signaling in COPD: Deciphering genetic and cellular susceptibilities for future therapeutic regimen. Swiss Med. Wkly. 2009, 139, 554–563. [Google Scholar]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Kubo, H.; Ota, C.; Takahashi, T.; Tando, Y.; Suzuki, T.; Fujino, N.; Makiguchi, T.; Takagi, K.; Suzuki, T.; et al. The increase of microRNA-21 during lung fibrosis and its contribution to epithelial-mesenchymal transition in pulmonary epithelial cells. Respir. Res. 2013, 14, 95. [Google Scholar] [CrossRef] [Green Version]
- Moimas, S.; Salton, F.; Kosmider, B.; Ring, N.; Volpe, M.C.; Bahmed, K.; Braga, L.; Rehman, M.; Vodret, S.; Graziani, M.L.; et al. miR-200 family members reduce senescence and restore idiopathic pulmonary fibrosis type II alveolar epithelial cell transdifferentiation. ERJ Open Res. 2019, 5, 00138-2019. [Google Scholar] [CrossRef] [Green Version]
- Rice, W.R.; Conkright, J.J.; Na, C.L.; Ikegami, M.; Shannon, J.M.; Weaver, T.E. Maintenance of the mouse type II cell phenotype in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L256–L264. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 213–224. [Google Scholar] [PubMed] [Green Version]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Flieger, D.; Hoff, A.S.; Sauerbruch, T.; Schmidt-Wolf, I.G. Influence of cytokines, monoclonal antibodies and chemotherapeutic drugs on epithelial cell adhesion molecule (EpCAM) and LewisY antigen expression. Clin. Exp. Immunol. 2001, 123, 9–14. [Google Scholar] [CrossRef]
- Starlets, D.; Gore, Y.; Binsky, I.; Haran, M.; Harpaz, N.; Shvidel, L.; Becker-Herman, S.; Berrebi, A.; Shachar, I. Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood 2006, 107, 4807–4816. [Google Scholar] [CrossRef]
- Messier, E.M.; Mason, R.J.; Kosmider, B. Efficient and rapid isolation and purification of mouse alveolar type II epithelial cells. Exp. Lung Res. 2012, 38, 363–373. [Google Scholar] [CrossRef]
- Monici, M. Cell and tissue autofluorescence research and diagnostic applications. Biotechnol. Annu. Rev. 2005, 11, 227–256. [Google Scholar]
- Heikal, A.A. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark. Med. 2010, 4, 241–263. [Google Scholar] [CrossRef] [Green Version]
- Fujino, N.; Kubo, H.; Ota, C.; Suzuki, T.; Suzuki, S.; Yamada, M.; Takahashi, T.; He, M.; Suzuki, T.; Kondo, T.; et al. A novel method for isolating individual cellular components from the adult human distal lung. Am. J. Respir. Cell Mol. Biol. 2012, 46, 422–430. [Google Scholar] [CrossRef]
- Trzpis, M.; McLaughlin, P.M.; de Leij, L.M.; Harmsen, M.C. Epithelial cell adhesion molecule: More than a carcinoma marker and adhesion molecule. Am. J. Pathol. 2007, 171, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res. 2001, 2, 33–46. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Chen, H.; Chen, Z.; Liu, L. Hemoglobin is expressed in alveolar epithelial type II cells. Biochem. Biophys. Res. Commun. 2005, 333, 1348–1352. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.L.; Newton, D.A.; Spyropoulos, D.D.; Baatz, J.E. Hypoxia up-regulates expression of hemoglobin in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Teisanu, R.M.; Lagasse, E.; Whitesides, J.F.; Stripp, B.R. Prospective isolation of bronchiolar stem cells based upon immunophenotypic and autofluorescence characteristics. Stem Cells 2009, 27, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Ghaedi, M.; Le, A.V.; Hatachi, G.; Beloiartsev, A.; Rocco, K.; Sivarapatna, A.; Mendez, J.J.; Baevova, P.; Dyal, R.N.; Leiby, K.L.; et al. Bioengineered lungs generated from human iPSCs-derived epithelial cells on native extracellular matrix. J. Tissue Eng. Regen. Med. 2018, 12, e1623–e1635. [Google Scholar] [CrossRef]
- Ghaedi, M.; Calle, E.A.; Mendez, J.J.; Gard, A.L.; Balestrini, J.; Booth, A.; Bove, P.F.; Gui, L.; White, E.S.; Niklason, L.E. Human iPS cell-derived alveolar epithelium repopulates lung extracellular matrix. J. Clin. Investig. 2013, 123, 4950–4962. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Hata, H.; Ozawa, H.; Takata, K. Immunohistochemical localization of the aquaporins AQP1, AQP3, AQP4, and AQP5 in the mouse respiratory system. Acta Histochem. Cytochem. 2009, 42, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Krane, C.M.; Fortner, C.N.; Hand, A.R.; McGraw, D.W.; Lorenz, J.N.; Wert, S.E.; Towne, J.E.; Paul, R.J.; Whitsett, J.A.; Menon, A.G. Aquaporin 5-deficient mouse lungs are hyperresponsive to cholinergic stimulation. Proc. Natl. Acad. Sci. USA 2001, 98, 14114–14119. [Google Scholar] [CrossRef] [Green Version]
- Kreda, S.M.; Gynn, M.C.; Fenstermacher, D.A.; Boucher, R.C.; Gabriel, S.E. Expression and localization of epithelial aquaporins in the adult human lung. Am. J. Respir. Cell Mol. Biol. 2001, 24, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Flodby, P.; Borok, Z.; Banfalvi, A.; Zhou, B.; Gao, D.; Minoo, P.; Ann, D.K.; Morrisey, E.E.; Crandall, E.D. Directed expression of Cre in alveolar epithelial type 1 cells. Am. J. Respir. Cell Mol. Biol. 2010, 43, 173–178. [Google Scholar] [CrossRef] [Green Version]
- King, L.S.; Nielsen, S.; Agre, P. Aquaporins in complex tissues. I. Developmental patterns in respiratory and glandular tissues of rat. Am. J. Physiol. 1997, 273, C1541–C1548. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Deharvengt, S.; Marmarelis, M.; Korc, M. Concomitant targeting of EGF receptor, TGF-beta and SRC points to a novel therapeutic approach in pancreatic cancer. PLoS ONE 2012, 7, e39684. [Google Scholar] [CrossRef]
- Tian, Y.C.; Chen, Y.C.; Chang, C.T.; Hung, C.C.; Wu, M.S.; Phillips, A.; Yang, C.W. Epidermal growth factor and transforming growth factor-beta1 enhance HK-2 cell migration through a synergistic increase of matrix metalloproteinase and sustained activation of ERK signaling pathway. Exp. Cell Res. 2007, 313, 2367–2377. [Google Scholar] [CrossRef]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Konigshoff, M.; Jayachandran, A.; Handley, D.; Seeger, W.; Kaminski, N.; Eickelberg, O. Transgelin is a direct target of TGF-beta/Smad3-dependent epithelial cell migration in lung fibrosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 1778–1789. [Google Scholar]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116 Pt 2, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Alipio, Z.A.; Jones, N.; Liao, W.; Yang, J.; Kulkarni, S.; Sree Kumar, K.; Hauer-Jensen, M.; Ward, D.C.; Ma, Y.; Fink, L.M. Epithelial to mesenchymal transition (EMT) induced by bleomycin or TFG(b1)/EGF in murine induced pluripotent stem cell-derived alveolar Type II-like cells. Differ. Res. Biol. Divers. 2011, 82, 89–98. [Google Scholar]
- Ouyang, H.; Gore, J.; Deitz, S.; Korc, M. microRNA-10b enhances pancreatic cancer cell invasion by suppressing TIP30 expression and promoting EGF and TGF-beta actions. Oncogene 2017, 36, 4952. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Wang, F.; Gu, Y.; Pei, X.; Guo, G.; Yu, C.; Li, L.; Zhu, M.; Xiong, Y.; Wang, Y. MicroRNA-21 induces breast cancer cell invasion and migration by suppressing smad7 via EGF and TGF-beta pathways. Oncol. Rep. 2016, 35, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, H.; Liu, J.; Tu, X.; Zang, Y.; Zhu, J.; Chen, J.; Dong, L.; Zhang, J. miR-30 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting Snail1. Biochem. Biophys. Res. Commun. 2012, 417, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, M.; Lan, H.; Yu, X. miR-30a negatively regulates TGF-beta1-induced epithelial-mesenchymal transition and peritoneal fibrosis by targeting Snai1. Am. J. Pathol. 2013, 183, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Mestdagh, P.; Bostrom, A.K.; Impens, F.; Fredlund, E.; Van Peer, G.; De Antonellis, P.; von Stedingk, K.; Ghesquiere, B.; Schulte, S.; Dews, M.; et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-beta pathway in neuroblastoma. Mol. Cell 2010, 40, 762–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Shi, J.Y.; Zhu, G.Q.; Shi, B. MiR-17-92 cluster regulates cell proliferation and collagen synthesis by targeting TGFB pathway in mouse palatal mesenchymal cells. J. Cell. Biochem. 2012, 113, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Chen, F.; Wang, K.; Song, Y.; Fei, X.; Wu, B. miR-15a/miR-16 cluster inhibits invasion of prostate cancer cells by suppressing TGF-beta signaling pathway. Biomed. Pharmacother. 2018, 104, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Jackstadt, R.; Siemens, H.; Li, H.; Kirchner, T.; Hermeking, H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2014, 74, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [Green Version]
- Selman, M.; Lopez-Otin, C.; Pardo, A. Age-driven developmental drift in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2016, 48, 538–552. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, Y.; Xie, T.; Liu, N.; Chen, H.; Geng, Y.; Kurkciyan, A.; Mena, J.M.; Stripp, B.R.; Jiang, D.; et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 2016, 22, 1285–1293. [Google Scholar] [CrossRef]
- Marmai, C.; Sutherland, R.E.; Kim, K.K.; Dolganov, G.M.; Fang, X.; Kim, S.S.; Jiang, S.; Golden, J.A.; Hoopes, C.W.; Matthay, M.A.; et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L71–L78. [Google Scholar] [CrossRef]
- Chen, J.; Wang, L.; Matyunina, L.V.; Hill, C.G.; McDonald, J.F. Overexpression of miR-429 induces mesenchymal-to-epithelial transition (MET) in metastatic ovarian cancer cells. Gynecol. Oncol. 2011, 121, 200–205. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Hill, L.; Browne, G.; Tulchinsky, E. ZEB/miR-200 feedback loop: At the crossroads of signal transduction in cancer. Int. J. Cancer 2013, 132, 745–754. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Hu, X.; Macdonald, D.M.; Huettner, P.C.; Feng, Z.; El Naqa, I.M.; Schwarz, J.K.; Mutch, D.G.; Grigsby, P.W.; Powell, S.N.; Wang, X. A miR-200 microRNA cluster as prognostic marker in advanced ovarian cancer. Gynecol. Oncol. 2009, 114, 457–464. [Google Scholar] [CrossRef]
- Kurashige, J.; Kamohara, H.; Watanabe, M.; Hiyoshi, Y.; Iwatsuki, M.; Tanaka, Y.; Kinoshita, K.; Saito, S.; Baba, Y.; Baba, H. MicroRNA-200b regulates cell proliferation, invasion, and migration by directly targeting ZEB2 in gastric carcinoma. Ann. Surg. Oncol. 2012, 19 (Suppl. 3), S656–S664. [Google Scholar] [CrossRef]
- Zidar, N.; Bostjancic, E.; Gale, N.; Kojc, N.; Poljak, M.; Glavac, D.; Cardesa, A. Down-regulation of microRNAs of the miR-200 family and miR-205, and an altered expression of classic and desmosomal cadherins in spindle cell carcinoma of the head and neck--hallmark of epithelial-mesenchymal transition. Hum. Pathol. 2011, 42, 482–488. [Google Scholar] [CrossRef]
- Braun, J.; Hoang-Vu, C.; Dralle, H.; Huttelmaier, S. Downregulation of microRNAs directs the EMT and invasive potential of anaplastic thyroid carcinomas. Oncogene 2010, 29, 4237–4244. [Google Scholar] [CrossRef] [Green Version]
- Tellez, C.S.; Juri, D.E.; Do, K.; Bernauer, A.M.; Thomas, C.L.; Damiani, L.A.; Tessema, M.; Leng, S.; Belinsky, S.A. EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 2011, 71, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Hu, Q.; Hu, L.X.; Lin, X.R.; Liu, J.Q.; Lin, X.; Dinglin, X.X.; Zeng, J.Y.; Hu, H.; Luo, M.L.; et al. miR-200b regulates epithelial-mesenchymal transition of chemo-resistant breast cancer cells by targeting FN1. Discov. Med. 2017, 24, 75–85. [Google Scholar]
- Ebert, M.S.; Sharp, P.A. Roles for microRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Janakiraman, H.; House, R.P.; Gangaraju, V.K.; Diehl, J.A.; Howe, P.H.; Palanisamy, V. The Long (lncRNA) and Short (miRNA) of It: TGFbeta-Mediated Control of RNA-Binding Proteins and Noncoding RNAs. Mol. Cancer Res. 2018, 16, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Blahna, M.T.; Hata, A. Smad-mediated regulation of microRNA biosynthesis. FEBS Lett. 2012, 586, 1906–1912. [Google Scholar] [CrossRef] [Green Version]
- Butz, H.; Racz, K.; Hunyady, L.; Patocs, A. Crosstalk between TGF-beta signaling and the microRNA machinery. Trends Pharmacol. Sci. 2012, 33, 382–393. [Google Scholar] [CrossRef]
- Wang, D.; Haviland, D.L.; Burns, A.R.; Zsigmond, E.; Wetsel, R.A. A pure population of lung alveolar epithelial type II cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4449–4454. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Huang, W.; Xu, R.; Nie, Y.; Cao, X.; Meng, J.; Xu, X.; Hu, S.; Zheng, Z. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J. Cell. Mol. Med. 2012, 16, 2150–2160. [Google Scholar] [CrossRef]
- Xie, T.; Liang, J.; Guo, R.; Liu, N.; Noble, P.W.; Jiang, D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol. Genom. 2011, 43, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Pandit, K.V.; Milosevic, J.; Kaminski, N. MicroRNAs in idiopathic pulmonary fibrosis. Transl. Res. J. Lab. Clin. Med. 2011, 157, 191–199. [Google Scholar] [CrossRef]
- Dews, M.; Fox, J.L.; Hultine, S.; Sundaram, P.; Wang, W.; Liu, Y.Y.; Furth, E.; Enders, G.H.; El-Deiry, W.; Schelter, J.M.; et al. The myc-miR-17~92 axis blunts TGF{beta} signaling and production of multiple TGF{beta}-dependent antiangiogenic factors. Cancer Res. 2010, 70, 8233–8246. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, D.L. Transcriptional Regulation of microRNA Genes and the Regulatory Networks in Which They Participate. Ph.D. Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 2008. Available online: http://d-scholarship.pitt.edu/8802/8801/DLCorcoran_etd_081208.pdf (accessed on 11 June 2022).
- Zhao, L.; Yee, M.; O’Reilly, M.A. Transdifferentiation of alveolar epithelial type II to type I cells is controlled by opposing TGF-beta and BMP signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L409–L418. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies for Flow Cytometry and Cell Sorting (ITC: Isotype Control): | |||||
| Antigen | Host | Isotype | Fluorochrome | Clone | Company |
| CD31 | Rat | IgG2a, k | APC | MEC 13.3 | BD Pharmingen |
| ITC for CD31 | Rat | IgG2a, k | APC | R35-95 | BD Pharmingen |
| CD31 | Rat | IgG2a, k | PE | 390 | BioLegend |
| ITC for CD31 | Rat | IgG2a, k | PE | RTK2758 | BioLegend |
| CD45 | Rat | IgG2b, k | APC | 30-F11 | BD Pharmingen |
| ITC for CD45 | Rat | IgG2b, k | APC | A95-1 | BD Pharmingen |
| CD74 | Rat | IgG2b, k | FITC | In-1 | BD Pharmingen |
| ITC for CD74 | Rat | IgG2b, k | FITC | A95-1 | BD Pharmingen |
| Primary Antibodies for Immunofluorescence Staining: | |||||
| Antigen | Host | Isotype | Clone | Company | |
| Pan-cytokeratin | Goat | IgG1 | C-11 | Abcam | |
| E-Cadherin | Mouse | IgG2a, k | 36/E-Cadherin | BD Pharmingen | |
| Alpha-SMA | Mouse | IgG2a | 1A4 | Sigma | |
| CD31 | Rabbit | IgG | Polyclonal | Abcam | |
| Pro-SPC | Rabbit | IgG | Polyclonal | Chemicon/Millipore | |
| CCSP | Rabbit | IgG | Polyclonal | Upstate/Millipore | |
| CD45 | Rat | IgG2b, k | 30-F11 | BD Pharmingen | |
| Secondary Antibodies for Immunofluorescence Staining: | |||||
| Antigen | Host | Isotype | Fluorochrome | Company | |
| Rabbit-IgG (H+L) | Goat | IgG | Alexa Fluor 555 | Invitrogen | |
| Mouse-IgG (H+L) | Goat | IgG | Alexa Fluor 555 | Inivtrogen | |
| Rat-IgG (H+L) | Goat | IgG | Alexa Fluor 555 | Inivtrogen | |
| Goat-IgG (H+L) | Donkey | IgG | Alexa Fluor | Inivtrogen | |
| Gene Symbol | Species | NCBI GenBank Accession | Primers (5′->3′) | Product Size (bp) |
|---|---|---|---|---|
| Acta2 | Mmu | NM_007392 | Fwd: GCTGGTGATGATGCTCCCA Rev: GCCCATTCCAACCATTACTCC | 81 |
| Aqp5 | Mmu | NM_009701 | Fwd: CCTTATCCATTGGCTTGTCG Rev: CTGAACCGATTCATGACCAC | 115 |
| Cd74 | Mmu | NM_001042605 | Fwd: GATGGCTACTCCCTTGCTGA Rev: TGGGTCATGTTGCCGTACT | 93 |
| Cdh1 | Mmu | NM_009864 | Fwd: CCATCCTCGGAATCCTTGG Rev: TTTGACCACCGTTCTCCTCC | 89 |
| Hprt | Mmu | NM_013556 | Fwd: CCTAAGATGAGCGCAAGTTGAA Rev: CCACAGGACTAGAACACCTGCTAA | 86 |
| Pecam1 | Mmu | NM_008816 | Fwd: ATCGGCAAAGTGGTCAAGAG Rev: GGCATGTCCTTTTATGATCTCAG | 111 |
| Ptprc | Mmu | NM_001111316 | Fwd: GTCCCTACTTGCCTATGTCAATG Rev: CCGGGAGGTTTTCATTCC | 115 |
| Sftpa1 | Mmu | NM_023134 | Fwd: GGAGAGCCTGGAGAAAGGGGGC Rev: ATCCTTGCAAGCTGAGGACTCCC | 124 |
| Sftpc | Mmu | NM_011359 | Fwd: AGCAAAGAGGTCCTGATGGA Rev: GAGCAGAGCCCCTACAATCA | 153 |
| Tjp1 | Mmu | NM_009386 | Fwd: ACGAGATGCTGGGACTGACC Rev: AACCGCATTTGGCGTTACAT | 112 |
| ACTA2 | HSA | NM_001141945 | Fwd: GGCTCTGGGCTCTGTAAGG Rev: TTTGCTCTGTGCTTCGTCAC | 147 |
| BCL2 | HSA | NM_000633 | Fwd: CTGAGTACCTGAACCGGCA Rev: GAGAAATCAAACAGAGGCCG | 106 |
| BMPR2 | HSA | NM_001204 | Fwd: TGCCCTCCTGATTCTTGG Rev: CATAGCCGTTCTTGATTCTGC | 130 |
| CDH1 | HSA | NM_004360 | Fwd: ATACACTCTCTTCTCTCACGCTGTGT Rev: CATTCTGATCGGTTACCGTGATC | 89 |
| FN1 | HSA | NM_212482 | Fwd: CCGACCAGAAGTTTGGGTTCT Rev: CAATGCGGTACATGACCCCT | 81 |
| HPRT1 | HSA | NM_000194 | Fwd: TTGTTGTAGGATATGCCCTTGAC Rev: TCTCATCTTAGGCTTTGTATTTTGC | 105 |
| JUN | HSA | NM_002228 | Fwd: CAGAGAGACAGACTTGAGAACTTGAC Rev: GACGCAACCCAGTCCAAC | 100 |
| MAP2K4 | HSA | NM_003010 | Fwd: GGCCAAAGTATAAAGAGCTTCTGA Rev: CAGCGATATCAATCGACATACAT | 145 |
| RNA18S5 | HSA | NR_003286 | Fwd: GCAATTATTCCCCATGAACG Rev: AGGGCCTCACTAAACCATCC | 125 |
| RUNX2 | HSA | NM_001024630 | Fwd: TAGATGGACCTCGGGAACC Rev: GAGGCGGTCAGAGAACAAAC | 77 |
| SMAD3 | HSA | NM_005902 | Fwd: GTCAAGAGCCTGGTCAAGAAAC Rev: GATGGGACACCTGCAACC | 136 |
| SNAI1 | HSA | NM_005985 | Fwd: CTTCTCTAGGCCCTGGCTG Rev: AGGTTGGAGCGGTCAGC | 105 |
| TGFBR2 | HSA | NM_001024847 | Fwd: TCTGTGGATGACCTGGCTAAC Rev: TCATTTCCCAGAGCACCAG | 148 |
| TJP1 | HSA | NM_003257 | Fwd: GAGGAAACAGCTATATGGGAACAAC Rev: TGACGTTTCCCCACTCTGAAA | 120 |
| VIM | HSA | NM_003380 | Fwd: AGATGGCCCTTGACATTGAG Rev: TGAGTGGGTATCAACCAGAGG | 146 |
| Pathway Category | Pathways per Category | Examples of Pathways within Category |
|---|---|---|
| Cancer | 30 | Small and non-small cell lung cancer, p53 |
| Cellular growth, proliferation and development | 28 | PI3K/Akt, ILK, TGF-β, Integrin, FAK, mTOR |
| Cytokine signaling | 27 | Chemokine, IL-6, IL-8, IL-9, IL-10, IL-15, IL-17, IL-22, TNFR1 |
| Cellular immune response | 22 | CXCR4, HMGB1, NF-κB, dendritic cell maturation |
| Growth factor signaling | 21 | IGF-1, EGF, GM-CSF, VEGF, FGF, PDGF |
| Apoptosis signaling | 16 | PTEN, death receptor, 14-3-3, JAK/Stat, tight junction signaling |
| Cell cycle regulation | 13 | G1/S checkpoint regulation, G2/M DNA damage checkpoint regulation |
| Intracellular and second messenger | 13 | Glucocorticoid receptor, ERK/MAPK, Rac, Rho, Gα12/13, PAK |
| Neurotransmitters and other nervous system signaling | 13 | Neuregulin, ErbB, Ephrin receptor, axonal guidance |
| Organismal growth and development | 13 | Stem cell pluripotency, HGF, BMP, Wnt/β-catenin |
| Disease-specific pathways | 9 | Hepatic fibrosis, rheumatoid arthritis, Huntington’s disease |
| Cardiovascular signaling | 7 | Cardiac hypertrophy, atherosclerosis, thrombin signaling |
| Cellular stress and injury | 6 | HMGB1, HIF1α, p70S6K |
| Humoral immune response | 5 | CD40, IL-4, B cell receptor signaling |
| Nuclear receptor signaling | 5 | PPARα/RXRα activation, PPAR, RAR activation, VDR/RXR activation |
| Pathogen-influenced | 3 | LPS-stimulated MAPK signaling |
| Transcriptional regulation | 2 | Role of NANOG and Oct4 in mammalian embryonic stem cell pluripotency |
| Xenobiotic metabolism | 1 | Aryl hydrocarbon receptor signaling |
| Metabolism of cofactors and vitamins | 1 | Nicotinate and nicotinamide metabolism |
| Metabolism of complex lipids | 1 | Inositol phosphate metabolism |
| miRNA | miRBase MIMAT ID | Number of Targets | Pubmed ID for Exp. Obs. Interaction | mRNA Target | Transduction Level, Molecular Type |
|---|---|---|---|---|---|
| Mmu-miR-22-3p | 0000531 | 1 | 19011694 | Bmp7 | Extracellular ligand, growth factor |
| Mmu-miR-29a-3p | 0000535 | 2 | 19342382 | Tgfb3 | |
| Mmu-miR-30c-5p | 0000514 | 3 | 18258830 | Acvr1 | Plasma membrane receptor, kinase |
| Mmu-miR-24-3p | 0000219 | 6 | 17906079 | Acvr1b | |
| Mmu-miR-210-3p | 0000658 | 1 | 19520079 | Acvr1b | |
| Mmu-miR-29a-3p | 0000535 | 2 | 19342382 | Acvr2a | |
| Mmu-miR-125a-5p | 0000135 | 1 | 19738052 | Bmpr1b | |
| Mmu-miR-19a-3p | 0000651 | 1 | 19390056 | Bmpr2 | |
| Mmu-miR-25-3p | 0000652 | 2 | 19390056 | Bmpr2 | |
| Mmu-miR-17-5p | 0000649 | 3 | 19390056 | Bmpr2 | |
| Mmu-miR-17-5p | 0000649 | 3 | 20709030 | Tgfbr2 | |
| Mmu-miR-18a-3p | 0004626 | 1 | 19372139 | Kras | Cytoplasmatic signaling, enzyme |
| Mmu-miR-181a-5p | 0000210 | 2 | 20080834 | Kras | |
| Mmu-miR-16-5p | 0000527 | 4 | 20065103 | Map2k1 | Cytoplasmatic signaling, kinase |
| Mmu-miR-16-5p | 0000527 | 4 | 19861690 | Map2k4 | |
| Mmu-miR-24-3p | 0000219 | 6 | 19861690 | Map2k4 | |
| Mmu-miR-25-3p | 0000652 | 2 | 19861690 | Map2k4 | |
| Mmu-miR-24-3p | 0000219 | 6 | 19502786 | Mapk14 | |
| Mmu-miR-7a-5p | 0000677 | 2 | 19072608 | Raf1 | |
| Mmu-miR-199a-3p | 0000230 | 1 | 19251704 | Smad1 | Transcription factor |
| Mmu-miR-23b-3p | 0000125 | 3 | 19582816 | Smad3 | |
| Mmu-miR-24-3p | 0000219 | 6 | 19582816 | Smad3 | |
| Mmu-miR-27a-3p | 0000537 | 3 | 19582816 | Smad3 | |
| Mmu-miR-140-5p | 0000151 | 1 | 20071455 | Smad3 | |
| Mmu-miR-23b-3p | 0000125 | 3 | 19582816 | Smad4 | |
| Mmu-miR-24-3p | 0000219 | 6 | 19582816 | Smad4 | |
| Mmu-miR-27a-3p | 0000537 | 3 | 19582816 | Smad4 | |
| Mmu-miR-23b-3p | 0000125 | 3 | 19582816 | Smad5 | |
| Mmu-miR-24-3p | 0000219 | 6 | 19582816 | Smad5 | |
| Mmu-miR-27a-3p | 0000537 | 3 | 19582816 | Smad5 | |
| Mmu-miR-7a-5p | 0000677 | 2 | 17028171 | Fos | |
| Mmu-miR-222-3p | 0000670 | 1 | 20299489 | Fos | |
| Mmu-miR-16-5p | 0000527 | 4 | 18362358 | Jun | |
| Mmu-miR-30c-5p | 0000514 | 3 | 18668040 | Jun | |
| Mmu-miR-30c-5p | 0000514 | 3 | 21628588 | Runx2 | |
| Mmu-miR-218-5p | 0000663 | 1 | 21628588 | Runx2 | |
| Mmu-miR-16-5p | 0000527 | 4 | 18449891 | Bcl2 | Transcription factor target, transporter |
| Mmu-miR-17-5p | 0000649 | 3 | 19666108 | Bcl2 | |
| Mmu-miR-181a-5p | 0000210 | 2 | 20204284 | Bcl2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dehmel, S.; Weiss, K.J.; El-Merhie, N.; Callegari, J.; Konrad, B.; Mutze, K.; Eickelberg, O.; Königshoff, M.; Krauss-Etschmann, S. microRNA Expression Profile of Purified Alveolar Epithelial Type II Cells. Genes 2022, 13, 1420. https://doi.org/10.3390/genes13081420
Dehmel S, Weiss KJ, El-Merhie N, Callegari J, Konrad B, Mutze K, Eickelberg O, Königshoff M, Krauss-Etschmann S. microRNA Expression Profile of Purified Alveolar Epithelial Type II Cells. Genes. 2022; 13(8):1420. https://doi.org/10.3390/genes13081420
Chicago/Turabian StyleDehmel, Stefan, Katharina J. Weiss, Natalia El-Merhie, Jens Callegari, Birte Konrad, Kathrin Mutze, Oliver Eickelberg, Melanie Königshoff, and Susanne Krauss-Etschmann. 2022. "microRNA Expression Profile of Purified Alveolar Epithelial Type II Cells" Genes 13, no. 8: 1420. https://doi.org/10.3390/genes13081420
APA StyleDehmel, S., Weiss, K. J., El-Merhie, N., Callegari, J., Konrad, B., Mutze, K., Eickelberg, O., Königshoff, M., & Krauss-Etschmann, S. (2022). microRNA Expression Profile of Purified Alveolar Epithelial Type II Cells. Genes, 13(8), 1420. https://doi.org/10.3390/genes13081420