
Integrative Investigation of Root-Related mRNAs, lncRNAs and circRNAs of “Muscat Hamburg” (Vitis vinifera L.) Grapevine in Response to Root Restriction through Transcriptomic Analyses
 ,
,
Abstract
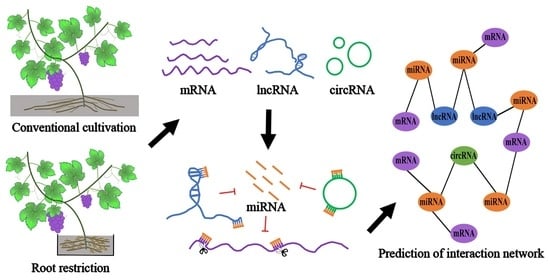
:
1. Introduction
2. Results
2.1. Root Phenotype under Conventional and Root-Restricted Cultivation
2.2. Sequencing Statistics in Different Roots Samples
2.3. Identification and Characterization of lncRNAs and circRNAs
2.4. Differential Expression Analyses of mRNA, lncRNAs, and circRNAs
2.5. Functional Enrichment Analyses
2.6. Experimental Validation of the circRNA Candidates
2.7. Real-Time Quantitative PCR (RT-qPCR)
2.8. CeRNA Network Analyses
3. Discussion
4. Materials and Methods
4.1. Plant Material and Treatments
4.2. Library Construction and Illumina Sequencing
4.3. Data Preprocessing and Genomic Alignment
4.4. Transcript Splicing, lncRNA Prediction, and Gene Quantification
4.5. Differential Expression Analysis and Functional Analysis
4.6. CircRNA Prediction, Expression Analysis and Interaction Research
4.7. Validation of circRNA in Grapevine
4.8. RT-qPCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, S.P.; Okamoto, G.; Hirano, K.; Lu, J.; Zhang, C.X. Effects of Restricted Rooting Volume on Vine Growth and Berry Development of Kyoho Grapevines. Am. J. Enol. Vitic. 2001, 52, 248–253. [Google Scholar]
- Eissenstat, D.M.; Yanai, R.D. The Ecology of Root Lifespan. Adv. Ecol. Res. 1997, 27, 1–60. [Google Scholar] [CrossRef]
- Chen, Q.; Deng, B.; Gao, J.; Zhao, Z.; Chen, Z.; Song, S.; Wang, L.; Zhao, L.; Xu, W.; Zhang, C.; et al. Comparative Analysis of miRNA Abundance Revealed the Function of Vvi-miR828 in Fruit Coloring in Root Restriction Cultivation Grapevine (Vitis vinifera L.). Int. J. Mol. Sci. 2019, 20, 4058. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gao, Z.; Zahid, M.S.; Li, D.; Javed, H.U.; Wang, L.; Song, S.; Zhao, L.; Xu, W.; Zhang, C.; et al. Small RNA Sequencing Analysis of miRNA Expression Reveals Novel Insihts into Root Formation under Root Restriction Cultivation in Grapevine (Vitis vinifera L.). Int. J. Mol. Sci. 2020, 21, 3513. [Google Scholar] [CrossRef]
- Leng, F.; Cao, J.; Wang, S.; Jiang, L.; Li, X.; Sun, C. Transcriptomic Analyses of Root Restriction Effects on Phytohormone Content and Signal Transduction during Grape Berry Development and Ripening. Int. J. Mol. Sci. 2018, 19, 2300. [Google Scholar] [CrossRef]
- Pires, R.C.; Furlani, P.R.; Sakai, E.; Lourenção, A.L.; Silva, E.A.; Neto, A.T.; Melo, A.M. Desenvolvimento e produtividade do tomateiro sob diferentes freqüências de irrigação em estufa. Hortic. Bras. 2009, 27, 228–234. [Google Scholar] [CrossRef]
- Mugnai, S.; Al-Debei, H.S. Growth reduction in root-restricted tomato plants is linked to photosynthetic impairment and starch accumulation in the leaves. Adv. Hortic. Sci. 2011, 25, 99–105. [Google Scholar] [CrossRef]
- Zakaria, N.I.; Ismail, M.R.; Awang, Y.; Megat Wahab, P.E.; Berahim, Z. Effect of Root Restriction on the Growth, Photosynthesis Rate, and Source and Sink Relationship of Chilli (Capsicum annuum L.) Grown in Soilless Culture. Biomed. Res. Int. 2020, 2020, 2706937. [Google Scholar] [CrossRef]
- Ismail, M.R.; Noor, K.M. Growth, water relations and physiological processes of starfruit (Averrhoa carambola L) plants under root growth restriction. Sci. Hortic. 1996, 66, 51–58. [Google Scholar] [CrossRef]
- Chekanova, J.A.; Gregory, B.D.; Reverdatto, S.V.; Chen, H.; Kumar, R.; Hooker, T.; Yazaki, J.; Li, P.; Skiba, N.; Peng, Q.; et al. Genome-wide high-resolution mapping of exosome substrates reveals hidden features in the Arabidopsis transcriptome. Cell 2007, 131, 1340–1353. [Google Scholar] [CrossRef]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA Maps Reveal New RNA Classes and a Possible Function for Pervasive Transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.Z.; Liu, M.; Zhao, M.G.; Chen, R.; Zhang, W.H. Identification and characterization of long non-coding RNAs involved in osmotic and salt stress in Medicago truncatula using genome-wide high-throughput sequencing. BMC Plant Biol. 2015, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Z.; Jiang, J.; Xu, C.; Kang, J.; Xiao, L.; Wu, M.; Xiong, J.; Guo, X.; Liu, H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell 2013, 25, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef]
- Taulli, R.; Loretelli, C.; Pandolfi, P.P. From pseudo-ceRNAs to circ-ceRNAs: A tale of cross-talk and competition. Nat. Struct. Mol. Biol. 2013, 20, 541–543. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Andres-Leon, E.; Nunez-Torres, R.; Rojas, A.M. miARma-Seq: A comprehensive tool for miRNA, mRNA and circRNA analysis. Sci. Rep. 2016, 6, 25749. [Google Scholar] [CrossRef]
- Ye, C.Y.; Chen, L.; Liu, C.; Zhu, Q.H.; Fan, L. Widespread noncoding circular RNAs in plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- St. Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef]
- Fan, C.; Hao, Z.; Yan, J.; Li, G. Genome-wide identification and functional analysis of lincRNAs acting as miRNA targets or decoys in maize. BMC Genom. 2015, 16, 793. [Google Scholar] [CrossRef]
- Yan, L.; Fan, G.; Li, X. Genome-wide analysis of three histone marks and gene expression in Paulownia fortunei with phytoplasma infection. BMC Genom. 2019, 20, 234. [Google Scholar] [CrossRef]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen, X.; Mu, M.; Wang, J.; Wang, X.; Wang, D.; Yin, Z.; Fan, W.; Wang, S.; Guo, L.; et al. Genome-Wide Analysis of Long Noncoding RNAs and Their Responses to Drought Stress in Cotton (Gossypium hirsutum L.). PLoS ONE 2016, 11, e0156723. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef]
- Chen, G.; Cui, J.; Wang, L.; Zhu, Y.; Lu, Z.; Jin, B. Genome-Wide Identification of Circular RNAs in Arabidopsis thaliana. Front. Plant Sci. 2017, 8, 1678. [Google Scholar] [CrossRef]
- Zhou, R.; Xu, L.; Zhao, L.; Wang, Y.; Zhao, T. Genome-wide identification of circRNAs involved in tomato fruit coloration. Biochem. Biophys. Res. Commun. 2018, 499, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Cheng, Y.; Zhang, C.; You, Q.; Shen, X.; Guo, W.; Jiao, Y. Genome-wide identification and characterization of circular RNAs by high throughput sequencing in soybean. Sci. Rep. 2017, 7, 5636. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Cui, L.; Zhou, Y.; Zhu, C.; Fan, D.; Gong, H.; Zhao, Q.; Zhou, C.; Zhao, Y.; Lu, D.; et al. Transcriptome-wide investigation of circular RNAs in rice. RNA 2015, 21, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, D.; Zirkel, A.; Kurian, L. Characterization of Circular RNAs (circRNA) Associated with the Translation Machinery. Methods Mol. Biol. 2018, 1754, 159–166. [Google Scholar] [CrossRef]
- Su, X.; Xing, J.; Wang, Z.; Chen, L.; Cui, M.; Jiang, B. microRNAs and ceRNAs: RNA networks in pathogenesis of cancer. Chin. J. Cancer Res. 2013, 25, 235–239. [Google Scholar] [CrossRef]
- Pacak, A.; Barciszewska-Pacak, M.; Swida-Barteczka, A.; Kruszka, K.; Sega, P.; Milanowska, K.; Jakobsen, I.; Jarmolowski, A.; Szweykowska-Kulinska, Z. Heat Stress Affects Pi-related Genes Expression and Inorganic Phosphate Deposition/Accumulation in Barley. Front. Plant Sci. 2016, 7, 926. [Google Scholar] [CrossRef]
- Xu, X.W.; Zhou, X.H.; Wang, R.R.; Peng, W.L.; An, Y.; Chen, L.L. Functional analysis of long intergenic non-coding RNAs in phosphate-starved rice using competing endogenous RNA network. Sci. Rep. 2016, 6, 20715. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, M.; Xing, L.; Li, W.; Jiang, H.; Wang, L.; Xu, M. Transcriptomic Analysis of Long Non-Coding RNAs and Coding Genes Uncovers a Complex Regulatory Network That Is Involved in Maize Seed Development. Genes 2017, 8, 274. [Google Scholar] [CrossRef]
- Ren, G.J.; Fan, X.C.; Liu, T.L.; Wang, S.S.; Zhao, G.H. Genome-wide analysis of differentially expressed profiles of mRNAs, lncRNAs and circRNAs during Cryptosporidium baileyi infection. BMC Genom. 2018, 19, 356. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, P.; Chen, Q.; Wang, J.; Chen, M. Identification and characterization of ncRNA-associated ceRNA networks in Arabidopsis leaf development. BMC Genom. 2018, 19, 607. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiaoming, L.; Xiang, W.; Yanhui, L.; Shu, J.; Maling, G.; Qing, M. Analyzing the interactions of mRNAs, miRNAs, lncRNAs and circRNAs to predict competing endogenous RNA networks in glioblastoma. J. Neurooncol. 2018, 137, 493–502. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, C.; Wang, Z.; Yang, Z.; Chen, D.; Wu, Y. LncRNA expression profile and ceRNA analysis in tomato during flowering. PLoS ONE 2019, 14, e0210650. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.J.; Shen, J. Circular RNA participates in the carcinogenesis and the malignant behavior of cancer. RNA Biol. 2017, 14, 514–521. [Google Scholar] [CrossRef]
- Wang, J.W.; Wang, L.J.; Mao, Y.B.; Cai, W.J.; Xue, H.W.; Chen, X.Y. Control of root cap formation by MicroRNA-targeted auxin response factors in Arabidopsis. Plant Cell 2005, 17, 2204–2216. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.; Bussell, J.D.; Pacurar, D.I.; Schwambach, J.; Pacurar, M.; Bellini, C. Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 2009, 21, 3119–3132. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Kats, L.; Salmena, L.; Weiss, D.; Tan, S.M.; Ala, U.; Karreth, F.; Poliseno, L.; Provero, P.; Di Cunto, F.; et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 2011, 147, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Boualem, A.; Laporte, P.; Jovanovic, M.; Laffont, C.; Plet, J.; Combier, J.P.; Niebel, A.; Crespi, M.; Frugier, F. MicroRNA166 controls root and nodule development in Medicago truncatula. Plant J. 2008, 54, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Sorin, C.; Declerck, M.; Christ, A.; Blein, T.; Ma, L.; Lelandais-Briere, C.; Njo, M.F.; Beeckman, T.; Crespi, M.; Hartmann, C. A miR169 isoform regulates specific NF-YA targets and root architecture in Arabidopsis. New Phytol. 2014, 202, 1197–1211. [Google Scholar] [CrossRef]
- Zhang, M.; Su, H.; Gresshoff, P.M.; Ferguson, B.J. Shoot-derived miR2111 controls legume root and nodule development. Plant Cell Environ. 2021, 44, 1627–1641. [Google Scholar] [CrossRef]
- Oyama, T.; Shimura, Y.; Okada, K. The IRE gene encodes a protein kinase homologue and modulates root hair growth in Arabidopsis. Plant J. 2002, 30, 289–299. [Google Scholar] [CrossRef]
- Gao, R.; Wang, Y.; Gruber, M.Y.; Hannoufa, A. miR156/SPL10 Modulates Lateral Root Development, Branching and Leaf Morphology in Arabidopsis by Silencing AGAMOUS-LIKE 79. Front. Plant Sci. 2017, 8, 2226. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Frugis, G.; Colgan, D.; Chua, N.H. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 2000, 14, 3024–3036. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Finn, R.D.; Mistry, J.; Schuster-Bockler, B.; Griffiths-Jones, S.; Hollich, V.; Lassmann, T.; Moxon, S.; Marshall, M.; Khanna, A.; Durbin, R.; et al. Pfam: Clans, web tools and services. Nucleic Acids Res. 2006, 34, D247–D251. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.Y.; Zhou, Z.Y. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Roberts, A.; Pachter, L. Streaming fragment assignment for real-time analysis of sequencing. Nat. Methods 2013, 10, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S. Analysing RNA-Seq data with the DESeq package. Mol. Biol. 2010, 43, 1–17. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Nucleic Acids Res. 2013, 1303, 1–3. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Tang, Q.; He, J.; Li, L.; Yang, N.; Yu, S.; Wang, M.; Zhang, Y.; Lin, J.; Cui, T.; et al. RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res. 2022, 50, D326–D332. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2–∆∆CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Valid Bases | Q30 | GC |
|---|---|---|---|---|---|---|---|
| NR12_1 | 50.65 M | 7.60 G | 50.23 M | 7.27 G | 95.70% | 93.66% | 47.69% |
| NR12_2 | 39.34 M | 5.90 G | 39.03 M | 5.67 G | 96.12% | 93.64% | 47.31% |
| NR12_3 | 39.31 M | 5.90 G | 39.00 M | 5.66 G | 96.07% | 93.74% | 46.50% |
| NR7_1 | 50.00 M | 7.50 G | 49.58 M | 7.12 G | 95.00% | 94.25% | 46.90% |
| NR7_2 | 48.08 M | 7.21 G | 47.72 M | 6.89 G | 95.54% | 94.28% | 46.57% |
| NR7_3 | 37.01 M | 5.55 G | 36.74 M | 5.35 G | 96.38% | 93.62% | 46.85% |
| RR12_1 | 42.49 M | 6.37 G | 42.13 M | 6.13 G | 96.22% | 93.56% | 46.97% |
| RR12_2 | 60.20 M | 9.03 G | 59.79 M | 8.69 G | 96.20% | 93.85% | 47.20% |
| RR12_3 | 47.77 M | 7.16 G | 47.37 M | 6.84 G | 95.40% | 93.76% | 47.64% |
| Term | All | NR12_vs_NR7-Diff | RR12_vs_NR7-Diff | RR12_vs_NR12-Diff |
|---|---|---|---|---|
| mRNA | 26,588 | 2320 | 1864 | 2440 |
| lncRNA | 1971 | 176 | 173 | 137 |
| circRNA | 2615 | 16 | 17 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Li, H.; Zhang, L.; Song, Y.; He, J.; Xu, W.; Ma, C.; Ren, Y.; Liu, H. Integrative Investigation of Root-Related mRNAs, lncRNAs and circRNAs of “Muscat Hamburg” (Vitis vinifera L.) Grapevine in Response to Root Restriction through Transcriptomic Analyses. Genes 2022, 13, 1547. https://doi.org/10.3390/genes13091547
Liu J, Li H, Zhang L, Song Y, He J, Xu W, Ma C, Ren Y, Liu H. Integrative Investigation of Root-Related mRNAs, lncRNAs and circRNAs of “Muscat Hamburg” (Vitis vinifera L.) Grapevine in Response to Root Restriction through Transcriptomic Analyses. Genes. 2022; 13(9):1547. https://doi.org/10.3390/genes13091547
Chicago/Turabian StyleLiu, Jingjing, Hui Li, Lipeng Zhang, Yue Song, Juan He, Wenping Xu, Chao Ma, Yi Ren, and Huaifeng Liu. 2022. "Integrative Investigation of Root-Related mRNAs, lncRNAs and circRNAs of “Muscat Hamburg” (Vitis vinifera L.) Grapevine in Response to Root Restriction through Transcriptomic Analyses" Genes 13, no. 9: 1547. https://doi.org/10.3390/genes13091547
APA StyleLiu, J., Li, H., Zhang, L., Song, Y., He, J., Xu, W., Ma, C., Ren, Y., & Liu, H. (2022). Integrative Investigation of Root-Related mRNAs, lncRNAs and circRNAs of “Muscat Hamburg” (Vitis vinifera L.) Grapevine in Response to Root Restriction through Transcriptomic Analyses. Genes, 13(9), 1547. https://doi.org/10.3390/genes13091547