1. Introduction
The analysis of DNA mixtures containing relatives is increasingly requested in criminal cases [
1]. Because of this, many probabilistic genotyping (PG) systems now provide alternative LR propositions in which relatives can be assessed [
2]. In these instances (e.g., LR = father vs. hypothetical son), the LRs obtained are significantly decreased due to the common ancestry that must be taken into account (i.e., degree of shared DNA). This differs from the LR algorithms typically reported in forensic DNA analysis where the person-of interest (POI) is evaluated against a random unrelated individual within the population [
3]. This is sufficient in a majority of cases, but problems can arise when multiple donors to the same mixture are related or when a non-donor relative of a true donor is being considered as a POI leading to the need for those alternative LR propositions. One option is to utilize a unified LR, which considers both relatives as well as unrelated individuals within the population when calculating the LR [
4]. However, because the unified LR assumes > 99.99% of individuals within a population are unrelated, a small impact is often seen on the LRs obtained [
5].
The high level of allele sharing seen with familial mixtures can further increase difficulty in assessing the number of contributors (NOC) [
6]. This is especially concerning as it is well known that underestimating the NOC to a mixture can result in missed donors. Studies examining simulated complex mixtures with the GlobalFiler
TM amplification kit have demonstrated a probability of 86%, 61%, and 17% of 6-person, 5-person, and 4-person mixtures respectively appearing as N−1 or fewer contributors [
7]. While peak height was not accounted for in these mixtures, neither was dropout, or stutter masking. Other studies have reported even greater NOC underestimation rates with prepared complex mixtures: 100%, 64%, and 23% of 6-person, 5-person, and 4-person mixtures respectively [
8,
9]. For mixtures comprised of related individuals, even higher rates underestimating the true NOC are expected.
Previous work on the testing and evaluation of PG systems with complex mixtures which considered the alternative possibility of there being present a relative of one of the donors has demonstrated that non-donor first-degree relatives (FDR) such as a full sibling or parent/child to a true donor can sometimes result in inclusionary LR values [
10,
11], with some even providing “very strong support” LRs (>10
6) [
11]. A study conducted with STRmix
TM examined more complicated familial mixtures comprised of a father, mother, and child, concluding that conditioning the LR on a single parent as well as utilization of the system’s Mx priors function was required to achieve reasonable results [
6]. However, this study did not examine the false inclusion of other non-donor relatives, and furthermore, prior information is not always available in actual casework to allow for this conditioning or the user-informed Mx priors function. Other internal crime laboratory validation reports have demonstrated a higher false inclusion rate for relatives compared to unrelated individuals especially with low template samples [
12,
13,
14,
15,
16,
17]. Therefore, fully resolving and identifying each of the individuals present in familial mixtures and excluding related non-donors is an important goal of the mixture deconvolution process and can be of great investigative value. In an attempt to achieve this goal, separation and analysis of each of the individual donors via single cell analysis was carried out by a combination of direct single cell subsampling/enhanced DNA typing [
18] and probabilistic genotyping [
19] and applied to three complex familial 4-person mixtures resulting in a probative gain of LR for all donors, an accurate identification of the NOC, and elimination of false inclusions of their non-donor relatives.
2. Methods
2.1. Sample Collection
Buccal swabs were collected from members of three separate families, as well as from two unrelated individuals (U1, U2). Family 1 consisted of a mother (M), father (F), and two children (C1 and C2). Family 2 consisted of a mother (M), father (F), and three children (C1, C2, and C3). Family 3 consisted of 5 full blood siblings (S1, S2, S3, S4, and S5). For each volunteer, a sterile cotton swab was used to swab the inside of the mouth and cheek according to procedures approved by the University of Central Florida’s Institutional Review Board.
2.2. Familial Mixture Creation
Equal concentrations of DNA donor extracts from the above familial donors were combined to create desired 2- and 3-person mixtures for later analysis by standard approaches only (i.e., not DSCS). These mixtures consisted of: (i) a father/mother mixture; (ii) a father/child mixture; and (iii) a full sibling/full sibling mixture. Three different 3-person mixtures were also examined including: (i) a father/mother/unrelated individual mixture, (ii) a father/mother/child individual mixture, and (iii) a 3 full-sibling mixture.
Buccal swabs collected from each family were used to create three distinct 4-person mixtures for later use with DSCS as well as standard approaches. Mixture 1 was comprised of the father and mother donors from family 1 as well as 2 unrelated individuals (i.e., F-M-U1-U2). Mixture 2 was comprised of the father, mother, and a child of family 2 as well as 1 unrelated individual (i.e., F-M-C1-U). Mixture 3 was comprised of 4 siblings from family 3 (i.e., S1-S2-S3-S4).
To create each 4-person mixture, the previously collected buccal swabs were agitated in separate aliquots (per donor) of 300 μL TE−4 buffer. Each donor solution was then centrifuged at 300 RCF for 7 min to create an epithelial cell pellet. Without disturbing the cell pellet, the supernatant to each solution was discarded and the pellets resuspended with 300 μL of TE−4 buffer. The CountessTM II FL (ThermoFisher Scientific, Carlsbad, CA, USA) automated cell counter was then used to determine the cell concentration of each cell suspension. Equal concentrations of the desired donor cell suspensions were combined to create each desired mixture (e.g., mixture 1, mixture 2, or mixture 3). Cell suspensions and mixtures were stored at 4 °C.
2.3. Slide Creation
As previously reported, the DSCS approach requires the creation of Gel-Film
® microscope slides which the created cell mixtures are deposited on (thus later referred to as mixture slides) as well as a 3M
TM adhesive slide which contains adhesive later utilized in the cell collection process [
18,
19,
20,
21,
22,
23].
To create the Gel-Film® slides, Gel-Pak® Gel-Film® (WF, ×8 retention level) (Hayward, CA, USA) was attached to clean glass microscope slides by way of the film’s adhesive backing. The film’s clear protective covering was then removed, and 60 μL of a cell suspension mixture (e.g., M-F-U1-U2) was pipetted onto the slide and spread out with a sterile swab. The resulting mixture slide was then stained 1–2 min with Trypan Blue and gently rinsed with nuclease-free water. Mixture slides were allowed to air-dry.
The adhesive slide reservoir was created by attaching 3M
TM (Allied Electronics, Fort Worth, TX, USA) adhesive to a clean glass microscope slide by way of double-sided tape. The adhesive backing was removed and the slide stored in a desiccator until needed [
18,
19,
20,
21,
22,
23].
2.4. Cell Recovery
A Leica M205C stereomicroscope (190–240× magnification) was used to visualize cells. Cells were collected by way of a sterile tungsten needle which was first utilized to obtain a small ball of 3M
TM adhesive that was then used to adhere selected visualized cells from the mixture slides [
18,
19,
20,
21,
22,
23]. The needle with adhesive and cell(s) was then inserted into a sterile 0.2 mL PCR flat-cap tube containing either 5 μL PunchSolution
TM (Promega, Madison, WI, USA) or 1 μL casework direct lysis mixture (Promega, Madison, WI, USA) until the 3M
TM adhesive was observed to solubilize. Forty 1- and 2-cell subsamples were collected from 4-person mixtures 1 (M-F-U1-U2) and 2 (M-F-C1-U) and forty 1-cell subsamples were collected from the 4-person mixture 3 (S1-S2-S3-S4).
2.5. Direct Lysis/Autosomal Short Tandem Repeat (STR) Amplification of Cells
For mixtures 1 and 2, cells were collected directly into 5 μL PunchSolutionTM and incubated at 90 °C → 30 min until the lysis solution evaporated. Cells collected from mixture 3 were collected into a lysis mixture comprised of 1 μL casework direct lysis buffer and 0.025 μL 50X diluted 1-thioglycerol. Samples were then incubated at 70 °C → 10 min.
After cell lysis, the subsamples were amplified using the GlobalFiler
TM Express amplification kit (ThermoFisher Scientific, Carlsbad, CA, USA) with a reduced reaction volume and increased cycle number. The GlobalFiler
TM Express reaction mix was prepared consisting of 2 μL PCR mix, 2 μL primer mix, and 1 μL 5× AmpSolution
TM (Promega, Madison, WI, USA). Samples were amplified using a protocol of 95 °C → 1 min; 32 cycles: 94 °C → 3 sec, 60 °C → 30 sec; 60 °C → 8 min; 4 °C → hold. Positive (1 μL of diluted DNA Control 007 (31.25 pg/μL)) and negative amplification controls (0-cell samples and amplification blanks) were included in each amplification batch [
19,
23].
2.6. Donor Reference Samples and Bulk Mixtures
2.6.1. DNA Isolation and Quantitation
DNA extraction was conducted on reference buccal swabs and 60 μL of each mixture cell suspension using the AutoMate Express™ Forensic DNA Extraction System (ThermoFisher Scientific, Carlsbad, CA, USA). Each extraction set contained an extraction blank and was quantified with the Quantifiler® Duo DNA Quantification kit (ThermoFisher Scientific, Carlsbad, CA, USA) using the Applied Biosystems’ 7500 real-time PCR instrument (ThermoFisher Scientific, Carlsbad, CA, USA).
2.6.2. Autosomal STR Amplification (Reference Samples and Mixtures)
The GlobalFilerTM (ThermoFisher Scientific, Carlsbad, CA, USA) amplification kit was used to amplify DNA from reference and bulk mixtures samples. One nanogram of input DNA was targeted, and the amplification protocol used was: 95 °C → 1 min; 29 cycles: 94 °C → 10 sec, 59 °C → 90 sec; 60 °C → 10 min; 4 °C → hold. Each amplification contained a positive and negative amplification control.
2.7. PCR Product Detection
GlobalFilerTM or GlobalFilerTM Express amplified product (1 μL) was added to 9.5 μL Hi-Di™ formamide (ThermoFisher Scientific, Carlsbad, CA, USA) and 0.5 μL GeneScanTM 600 LIZ® size standard (ThermoFisher Scientific, Carlsbad, CA, USA). Samples were then injected on the Applied Biosystems’ 3500 Genetic Analyzer using POP-4TM polymer and Module J6 (15 s injection, 1.2 kV, 60 °C). GeneMapper v1.6 software (ThermoFisher Scientific, Carlsbad, CA, USA) was used for analysis.
2.8. Probabilistic Genotyping (PG)
2.8.1. Standard Bulk Mixture Probabilistic Genotyping
Probabilistic genotyping software STRmix™ v2.8 (Institute of Environmental Science and Research, Auckland, New Zealand) was previously validated for use with standard bulk mixtures and reference samples. Each mixture was examined both by conditioning the LR on a known donor (i.e.,
) and without conditioning (i.e.,
). The FBI Caucasian database was used for all allele frequencies in all mixture experiments [
19]. Various number of contributor (NOC) and sub-source LR propositions were examined such as traditional LRs (i.e., evaluating the POI against unrelated individuals in the population), specific relative LRs (i.e., evaluating the POI against a theoretical related individual in the population), and unified LRs (i.e., evaluating the POI against both related and unrelated individuals in the population). Additionally, known FDR non-donors were tested as the POI in the H
1 or H
p proposition to test for advantageous false inclusions.
When the degree of support for the inclusionary proposition based upon the returned LR is expressed as a qualitative verbal statement, the SWGDAM recommendations are followed [
24,
25]: LR 2–99 (“limited support”), LR 100–9999 (“moderate support”), LR 10,000–999,999 (“strong support”), LR
> 1,000,000 (“very strong support”).
The use of unified LR propositions within STRmix
TM requires population settings including the relevant population size and the average number of children per family. The U.S. Census data from 2019 was used to estimate the US Caucasian population of 250,446,756 (i.e., 328,238,523*0.763) [
26] and 4 was utilized as the average number of children per family [
5].
2.8.2. DSCS Probabilistic Genotyping
Previously validated probabilistic genotyping Software STRmix™ v2.8 for single (or few) cell STR analysis [
19,
23] was used to obtain high resolution single source DNA profiles from 1 or 2 cell subsamples. For the DSCS specificity studies and the complex mixture studies, each 1- or 2-cell single source subsample was run as a single source
. For the complex mixtures’ deconvolution, the top 6 subsamples that returned the highest inclusionary LRs (i.e., log (LR)
> 1) for a specific donor were used for replicate analysis.
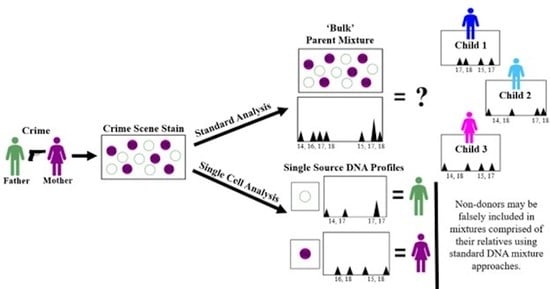
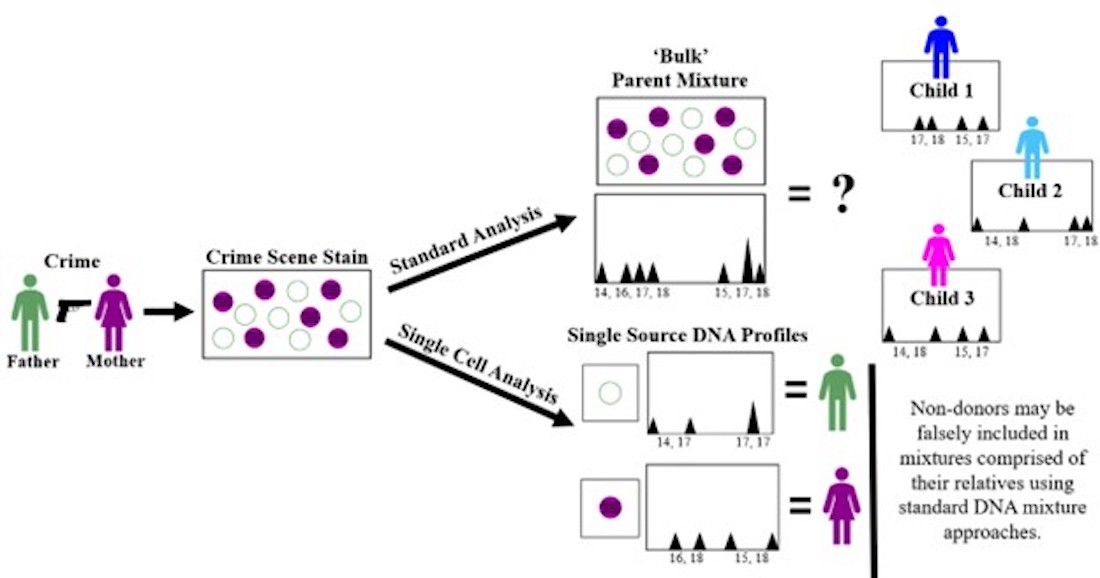
2.9. Description of DSCS Method
An infographic for the DSCS approach applied, as an example, to a 1:1 2-person father/mother mixture is provided in
Figure 1. Standard analysis of the mixed stain results in a mixed DNA profile (
Figure 1, left side). Non-donor children of the mother and father can then be falsely included as donors to the mixture (
Figure 1, bottom). Direct single cell subsampling of the same mixture allows for collection of 1–2 cell subsamples. This allows for single source profiles of the mother and father to be obtained from the mixture by 1-cell subsamples as well as some 2-cell subsamples. By increasing the number of cells collected in subsampling from 1 to 2, the amount of input DNA doubles thus increasing the probability of achieving a full profile if both cells originate from the same donor. However, some 2-cell subsamples still result in mixed profiles (i.e., 2-cell mini-mixtures) which can pose the same issues as standard familial mixtures (
Figure 1, right side). Therefore, it is recommended only single source subsamples be utilized with familial mixtures. The DSCS process referred to for convenience in this study not only encompasses single cell recovery and enhanced DNA typing (
Figure 1) but is combined with probabilistic genotyping using STRmix™ software (including the use of its replicate analysis functionality) [
19].
4. Discussion and Conclusions
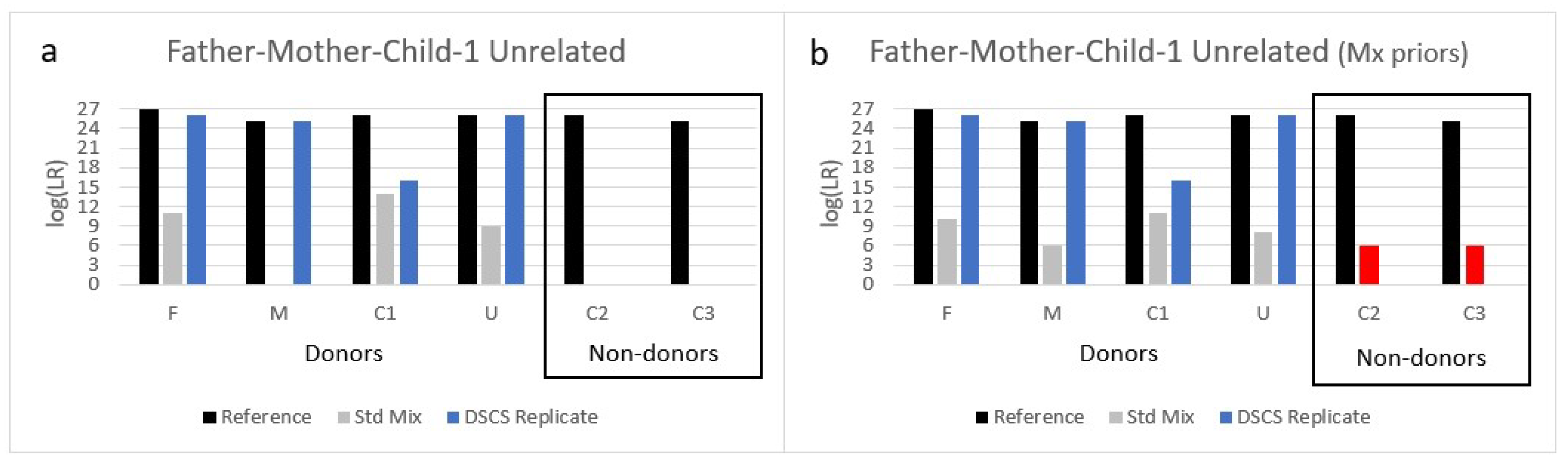
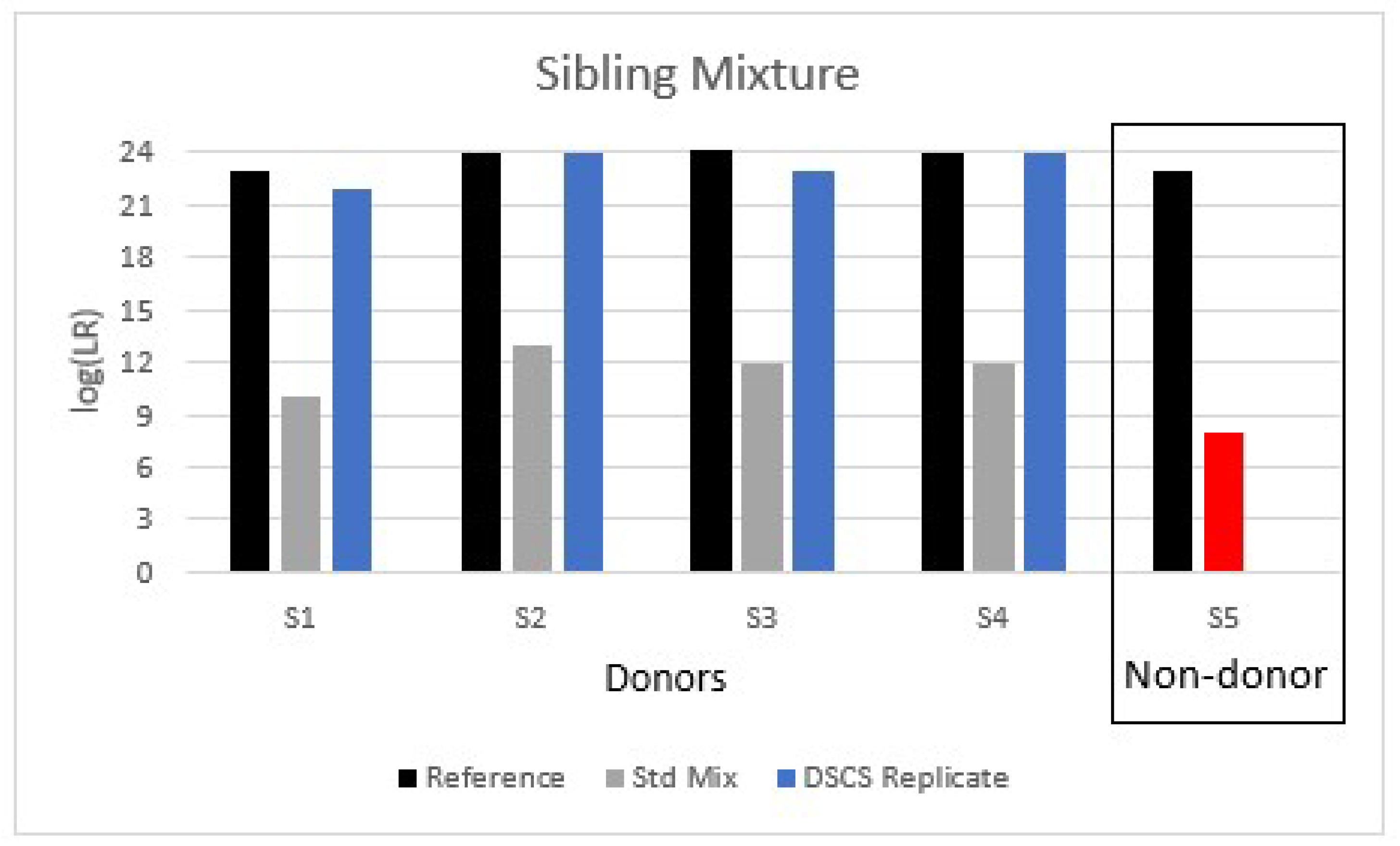
This small study of complex DNA mixtures in which two or more of the donors comprise FDRs confirms the need for DNA analysts to exercise caution when interpreting such mixtures. Most criminal cases will involve calculating an LR for the POI under an assumption that the exclusionary proposition (H2 or Hd) comprises individuals unrelated to the POI. However, as demonstrated in the current work, interpretation becomes more complicated if FDRs of the true donor POI are present in the mixture itself and another FDR, but who is not one of the mixture donors, becomes an alternative POI. There will arise casework situations where the analyst is blind to the fact that the mixture comprises FDR donors and that the presented suspect (POI) is not a donor but is an FDR of some of the true mixture donors. The analyst will likely process and interpret the mixture using a standard H2 = unrelated individuals interpretation scheme. We show here that such an approach with 2–4 person familial mixtures containing either the mother + father or multiple siblings can result in the false inclusion of FDRs (i.e., children or other siblings) with some LRs providing very strong support for the inclusionary hypothesis.
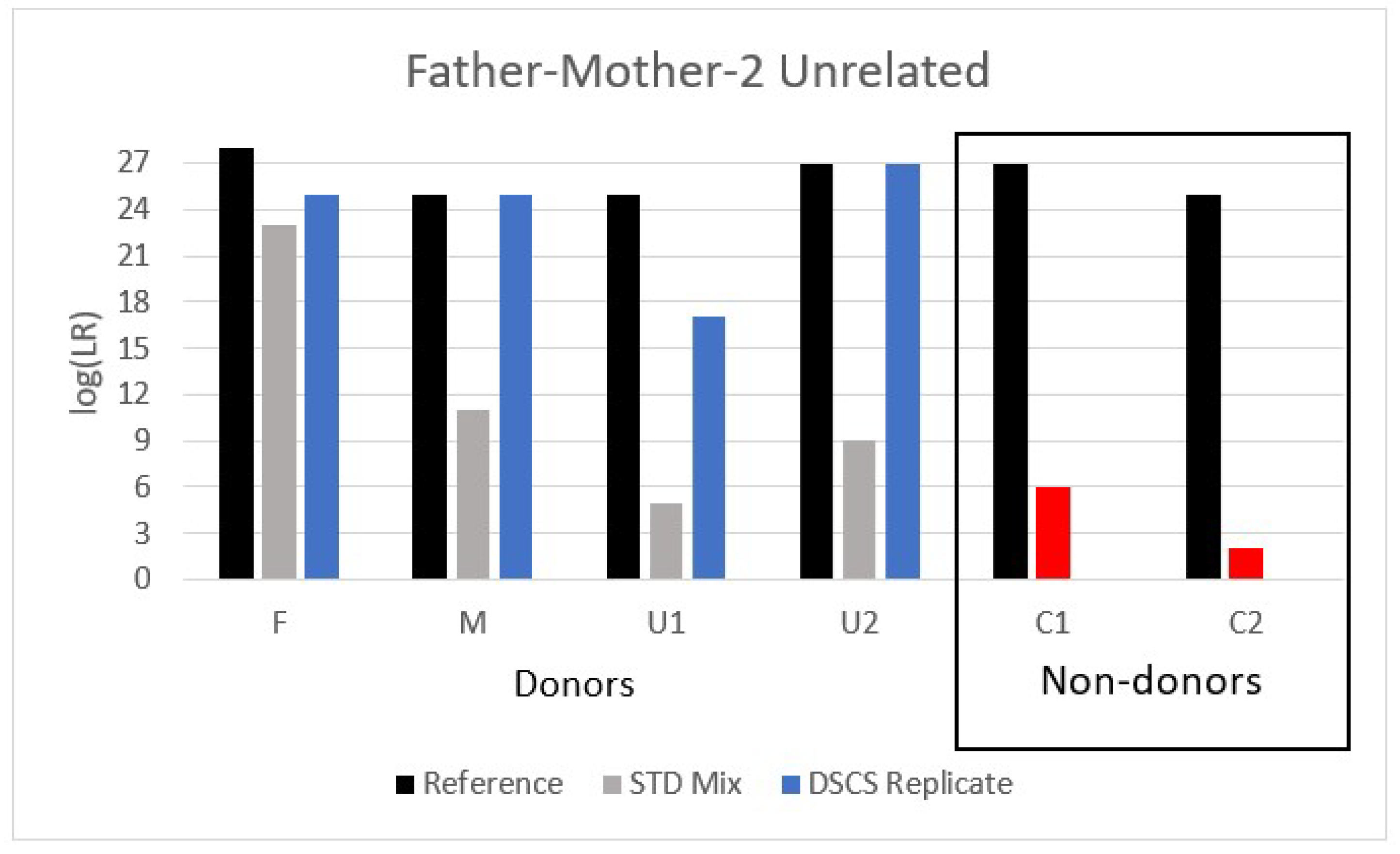
Although this situation of returning false positive LRs for non-donors in some familial mixtures is not ideal there are some potential PG software remedies available to the analyst to help ameliorate it, but these require additional contextual information about the case circumstances and justification for a modified LR calculation. Firstly, if the contextual information indicates that the assumption that there is a known donor present is objectively justifiable then then the mixture can be interpreted by conditioning it on the presence of that known donor. This effectively reduces the complexity of the mixture, especially if the known donor perchance is an FDR of the alternative POI (who is a true non-donor to the mixture), thereby further constraining the possible genotype combinations from the other donors. Although conditioning on the assumption that one of the known related donors was present can sometimes ameliorate this problem with 2–3 person familial mixtures, it can also reduce the degree of support for some of the other related donors present (
Table 2). Notably, of the three complex 4-person familial mixtures tested, two of them, despite conditioning, still returned false positive results for the FDR non-donors. Secondly, in addition to conditioning to reduce mixture complexity and constrain the genotype possibilities for other donors, different propositions were used to calculate a variety of different LRs for the 4-person mixtures including specific relative LRs (i.e., evaluating the POI against a related individual in the population), unified LRs (i.e., evaluating the POI against both related and unrelated individuals in the population), and the Mx priors function. None of these resulted in solving the problem of false inclusions of non-donor FDRs for all of the complex 4-person mixtures studied.
As expected, the high level of allele sharing seen with the type of complex familial mixtures studied here leads to an increased risk of underestimating the number of contributors (NOC) to a mixture. Indeed the 4-person complex mixtures studied could easily be misidentified as 3-person mixtures using the common method of electropherogram inspection for the maximum number of alleles detected at any locus. Interpreting all of these mixtures as 3-person mixtures by standard PG interpretation methods resulted in the false exclusion of true donors, including both FDRs and unrelated individuals.
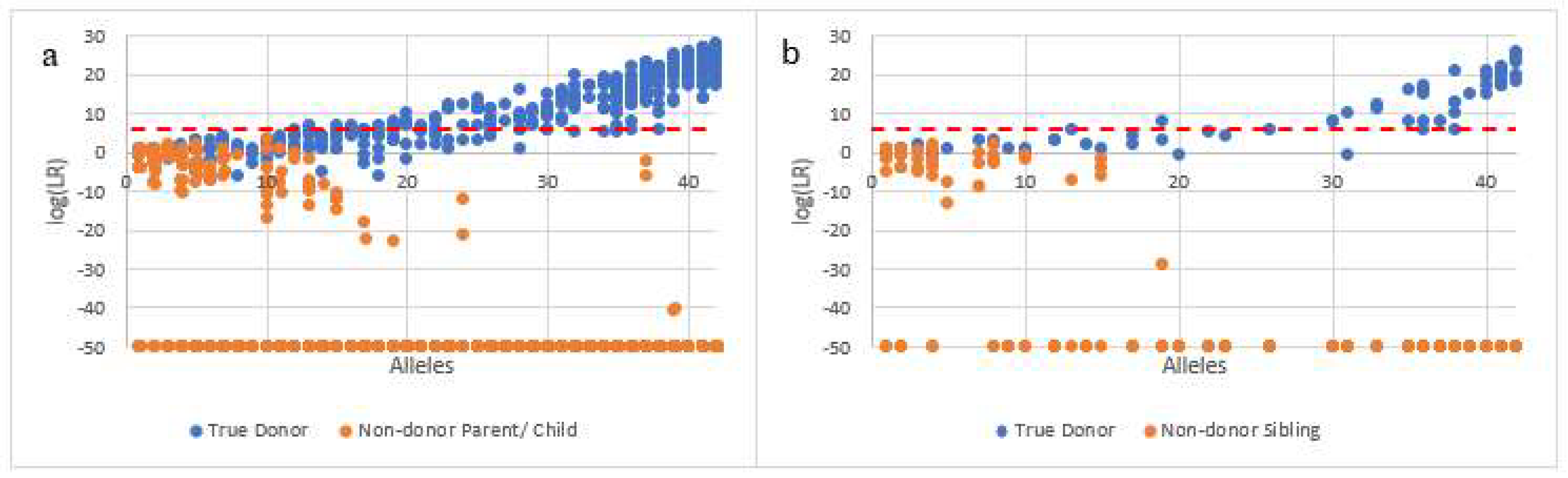
All of the above affirms that fully resolving and identifying each of the individuals present in familial mixtures and excluding related non-donors should be an important goal of the mixture deconvolution process. Although the goal is not always readily attainable, attempts should be made to ensure as much as possible that incorrect inclusion inferences are prevented or at least the strength of these false inclusions, as measured by LRs, is minimized. In order to try and achieve that goal, in the present work, instead of analyzing and interpreting complex familial mixtures via the standard bulk analysis approach, separation of each of the individual donors via single cell analysis was carried out by the DSCS process which, in this work, consisted of a combination of direct single cell subsampling, enhanced DNA typing and probabilistic genotyping. Once it was determined that single source cell subsamples could accurately distinguish known contributors from their first-degree relatives (
Figure 2), complex familial 4-person mixtures were analyzed using both standard bulk approaches and DSCS. We chose 4-person mixtures as they were some of the most complex mixture types that we could envision in a casework scenario (albeit not encountered on a routine basis) and that we could still analyze using standard PG approaches using the version of STRmix
TM (v2.8). By individually analyzing single cells collected from complex familial mixtures, full or near-full single-source DNA profiles were obtained for all true donors resulting in a probative gain of LR information for all donors, thus definitively implicating them as contributors to a mixture while the non-donor relatives were no longer falsely included. As the subsamples collected were single source, there was less risk in obtaining LRs that were disproportionately high or low as could be seen with complex familial mixtures using standard DNA mixture approaches. Furthermore, as familial mixtures have a high degree of allele sharing leading to an underrepresentation of the true NOC, single cell analysis could, and did here, provide an additional way of estimating the NOC. In the present work, the correct NOC was obtained for all three mixtures (i.e., an NOC = 4) based on the number of different DNA profiles recovered by DSCS. Indeed, it is envisioned that the DSCS process
per se, upon the future development of statistical clustering methods based upon the number of distinguishable genotype-related clusters recoverable from a mixture, could be used to empirically and directly determine the mixture’s NOC.
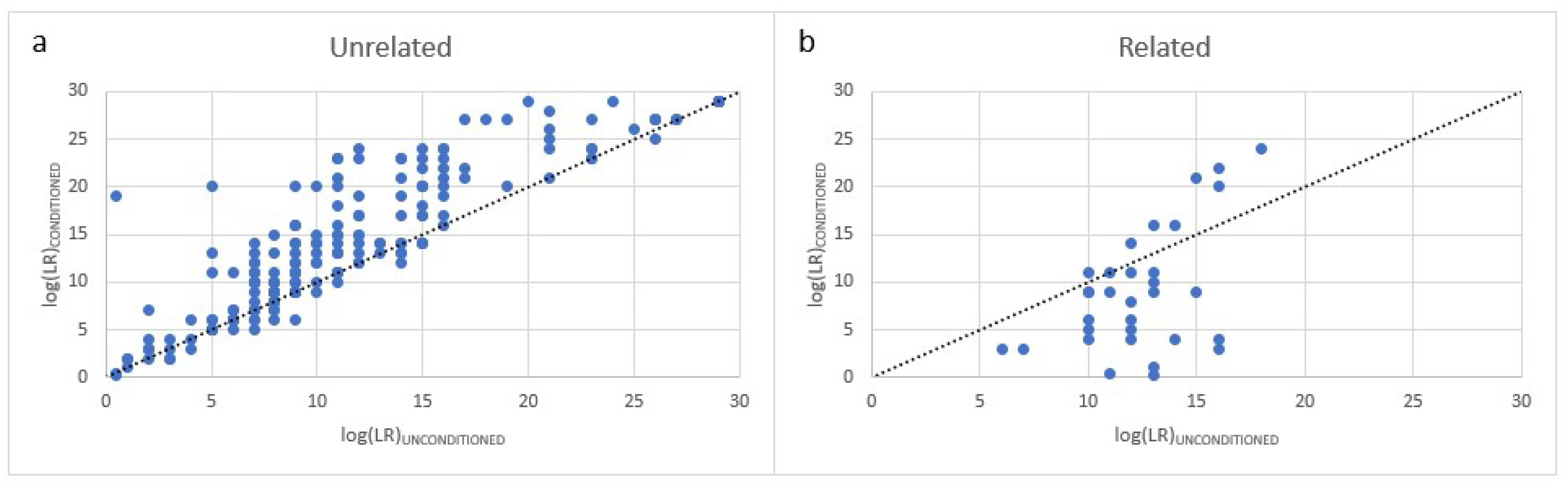
If case context does not identify the mother or father as a known donor, then DSCS could also provide a single source DNA profile for them allowing the standard bulk mixture to be conditioned on their inclusion. Such peeling typically results in an improvement in the LR recovery for a mixture comprising unrelated individuals [
27]. However as demonstrated in this study a decrease in the LR obtained for an individual when conditioning is employed may provide an indication that related individuals are present within the mixture.
Finally, the implementation of DSCS into routine casework could be achieved using the methodology described herein since only very basic equipment found in most forensic biology laboratories is required. Nevertheless, more widespread implementation of a single cell subsampling, DNA typing and interpretation strategy for mixture analysis would be facilitated by the automation of the DSCS process instead of the manual cell recovery process described here. For example, a combined microfluidics separation and encapsulated digital-droplet single-cell amplification system [
28] designed for STR analysis could result in the complete deconvolution of all mixture components to their single source state, thus potentially recovering the complete genotype information present in the sample.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}