
Gene Regulation during Carapacial Ridge Development of Mauremys reevesii: The Development of Carapacial Ridge, Ribs and Scutes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. RNA Extraction, Library Construction and Sequencing, Data Quality Control and Basic Annotation
2.3. Differentially Expressed Genes
2.4. qRT-PCR
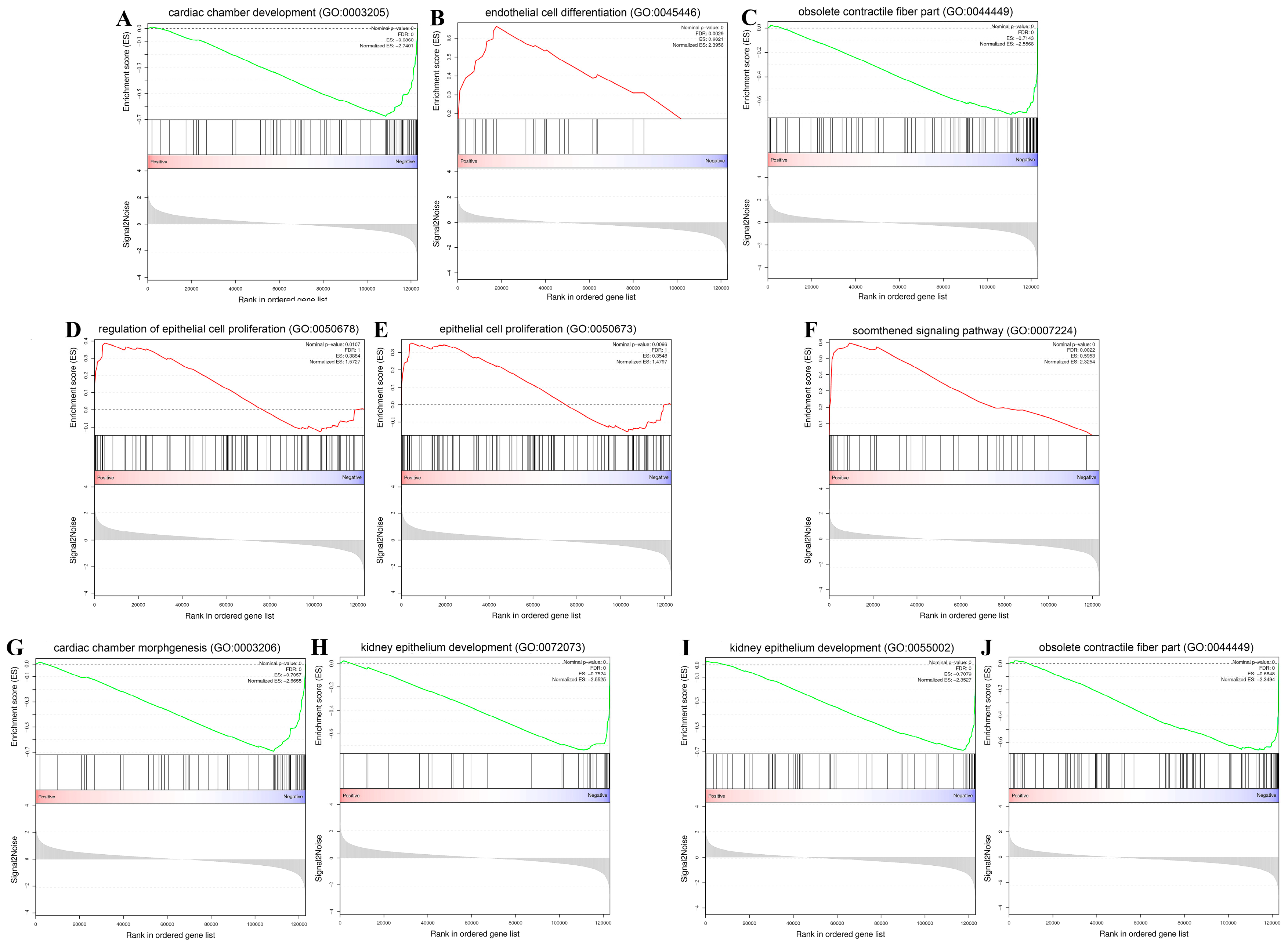
2.5. Gene Set Enrichment Analysis (GSEA)
2.6. Pathway Enrichment Analysis
2.7. Short Time-Series Expression Miner (STEM) Analysis
2.8. Constructing a Network of Key Genes
2.9. Weighted Gene Co-Expression Network Analysis (WGCNA)
3. Results
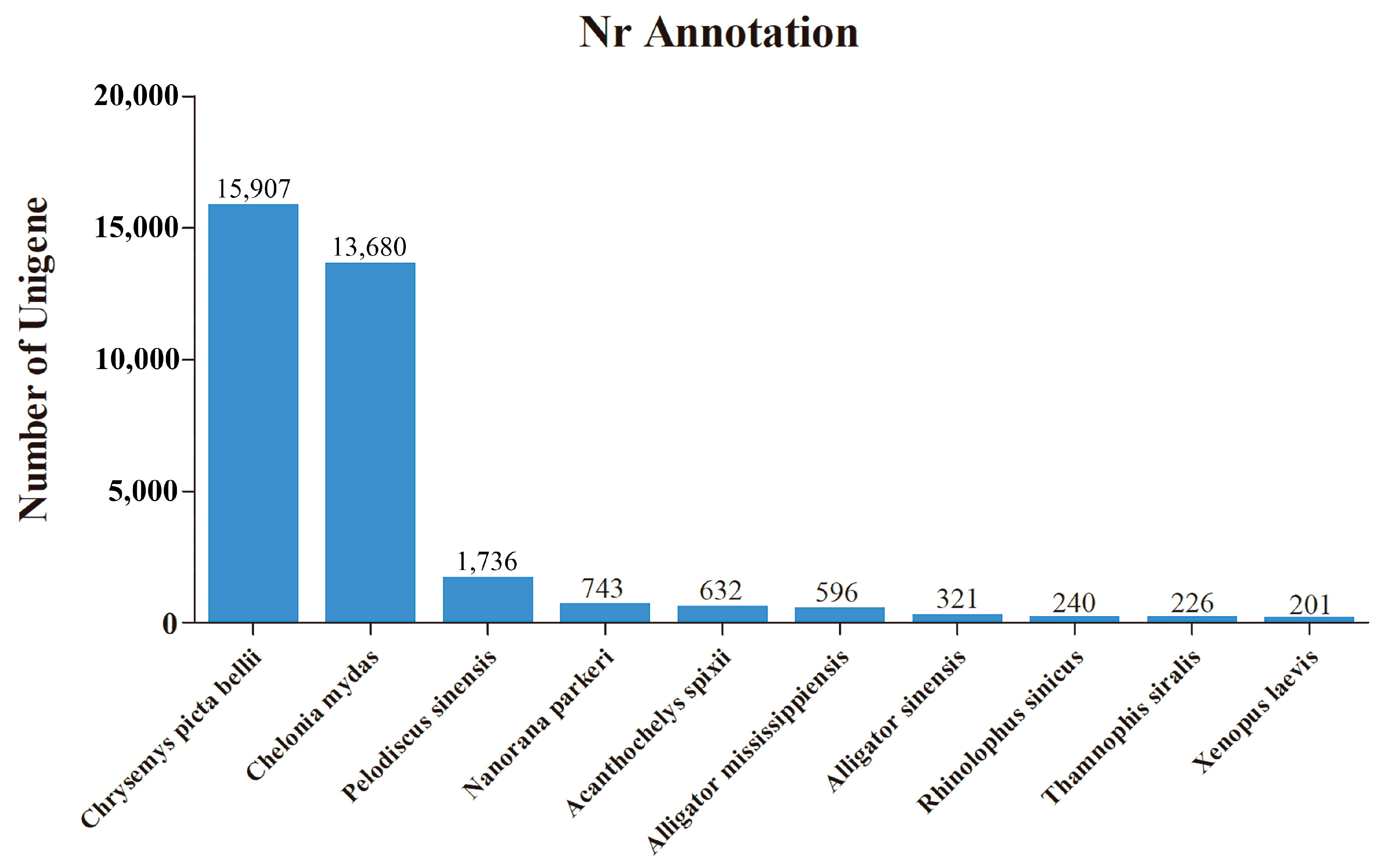
3.1. De Novo Transcriptome Assembly and Annotation
3.2. Analysis of Differentially Expressed Genes
3.3. KEGG Pathway Enrichment Analysis
3.4. DEGs Expression Trend Analysis
3.5. Real-Time RT-PCR Validation
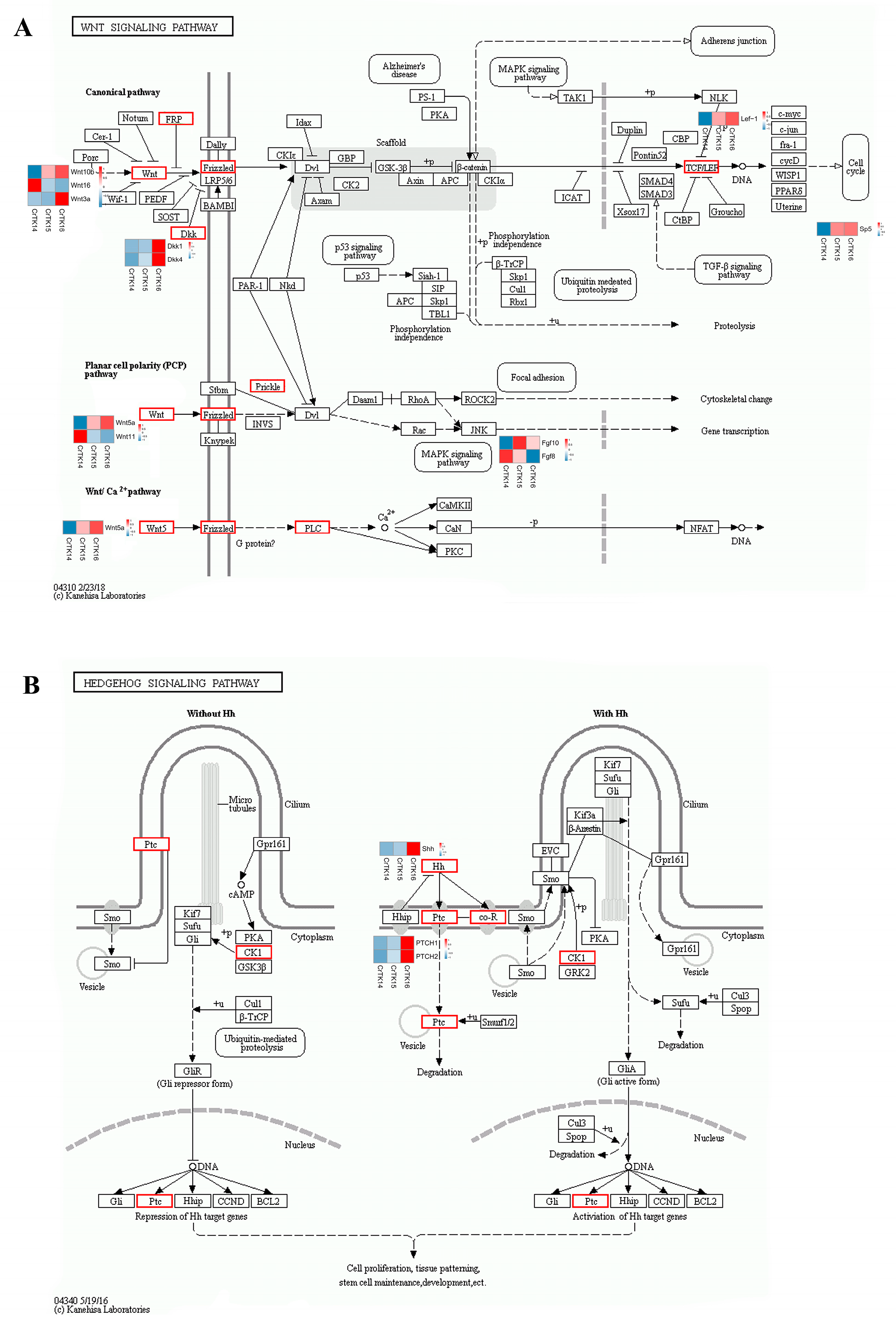
3.6. Interaction Network of Wnt Signaling Pathway
3.7. Weighted Co-Expression Network Analysis (WGCNA) of CR
3.8. Comparison and Analysis of Gene Modules Correlated with CR of M. reevesii and P. sinensis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Moustakas-Verho, J.E.; Cebra-Thomas, J.; Gilbert, S.F. Patterning of the turtle shell. Curr. Opin. Genet. Dev. 2017, 45, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Cordero, G.A.; Quinteros, K. Skeletal remodelling suggests the turtle’s shell is not an evolutionary straitjacket. Biol. Lett. 2015, 11, 20150022. [Google Scholar] [CrossRef] [PubMed]
- Kuratani, S.; Kuraku, S.; Nagashima, H. Evolutionary developmental perspective for the origin of turtles: The folding theory for the shell based on the developmental nature of the carapacial ridge. Evol. Dev. 2011, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cordero, G. Transcriptomic similarities and differences between the limb bud AER and unique carapacial ridge of turtle embryos. Evol Dev. 2020, 22, 370–383. [Google Scholar] [CrossRef]
- Nagashima, H.; Kuraku, S.; Uchida, K.; Kawashima-Ohya, Y.; Narita, Y.; Kuratani, S. Body plan of turtles: An anatomical, developmental and evolutionary perspective. Anat. Sci Int. 2012, 87, 1–13. [Google Scholar] [CrossRef]
- Kuraku, S.; Usuda, R.; Kuratani, S. Comprehensive survey of carapacial ridge-specific genes in turtle implies co-option of some regulatory genes in carapace evolution. Evol Dev. 2005, 7, 3–17. [Google Scholar] [CrossRef]
- Nagashima, H.; Kuraku, S.; Uchida, K.; Ohya, Y.; Narita, Y.; Kuratani, S. On the carapacial ridge in turtle embryos: Its developmental origin, function and the chelonian body plan. Development 2007, 134, 2219–2226. [Google Scholar] [CrossRef]
- Nagashima, H.; Shibata, M.; Taniguchi, M.; Ueno, S.; Kamezaki, N.; Sato, N. Comparative study of the shell development of hard- and soft-shelled turtles. J. Anat. 2014, 225, 60–70. [Google Scholar] [CrossRef]
- Nagashima, H.; Sugahara, F.; Takechi, M.; Ericsson, R.; Kawashima-Ohya, Y.; Narita, Y.; Kuratani, S. Evolution of the turtle body plan by the folding and creation of new muscle connections. Science 2009, 325, 193–196. [Google Scholar] [CrossRef]
- Schoch, R.R.; Sues, H.D. The origin of the turtle body plan: Evidence from fossils and embryos. Palaeontology 2020, 63, 375–393. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wu, X.C.; Rieppel, O.; Wang, L.T.; Zhao, L.J. An ancestral turtle from the Late Triassic of southwestern China. Nature 2008, 456, 497–501. [Google Scholar] [CrossRef]
- Schoch, R.R.; Sues, H.D. A Middle Triassic stem-turtle and the evolution of the turtle body plan. Nature 2015, 523, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fraser, N.C.; Rieppel, O.; Wu, X.C. A Triassic stem turtle with an edentulous beak. Nature 2018, 560, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.C.; Spinks, P.Q.; Shaffer, H.B. A global phylogeny of turtles reveals a burst of climate-associated diversification on continental margins. Proc. Natl. Acad. Sci. USA 2021, 118, e2012215118. [Google Scholar] [CrossRef] [PubMed]
- Alibardi, L. Proliferation in the epidermis of chelonians and growth of the horny scutes. J. Morphol. 2005, 265, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Moustakas-Verho, J.; Zimm, R.; Cebra-Thomas, J.; Lempiäinen, N.; Kallonen, A.; Mitchell, K.; Hämäläinen, K.; Salazar-Ciudad, I.; Jernvall, J.; Gilbert, S. The origin and loss of periodic patterning in the turtle shell. Development 2014, 141, 3033–3039. [Google Scholar] [CrossRef]
- Gaffney, E.S. The comparative osteology of the Triassic turtle Proganochelys. Bull. Am. Mus. Nat. Hist 1990, 194, 1–263. [Google Scholar]
- Sanchez-Villagra, M.R.; Muller, H.; Sheil, C.A.; Scheyer, T.M.; Nagashima, H.; Kuratani, S. Skeletal development in the Chinese soft-shelled turtle Pelodiscus sinensis (Testudines: Trionychidae). J. Morphol. 2009, 270, 1381–1399. [Google Scholar] [CrossRef]
- Burke, A.C. Development of the turtle carapace: Implications for the evolution of a novel bauplan. J. Morphol. 1989, 199, 363–378. [Google Scholar] [CrossRef]
- Burke, A.C. The Development and Evolution of the Turtle Body Plan, Inferring Intrinsic Aspects of the Evolutionary Process from Experimental Embryology. Am. Zool. 1991, 31, 616–627. [Google Scholar] [CrossRef]
- Moustakas, J. Development of the carapacial ridge: Implications for the evolution of genetic networks in turtle shell development. Evol. Dev. 2008, 10, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pascual-Anaya, J.; Zadissa, A.; Li, W.; Niimura, Y.; Huang, Z.; Li, C.; White, S.; Xiong, Z.; Fang, D.; et al. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat. Genet. 2013, 45, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Anaya, J.; Hirasawa, T.; Sato, I.; Kuraku, S.; Kuratani, S. Comparative analysis of pleurodiran and cryptodiran turtle embryos depicts the molecular ground pattern of the turtle carapacial ridge. Int. J. Dev Biol. 2014, 58, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, P.; Zhao, Y.; Zhou, Q.; Yang, J.; Hu, Q.; Liu, T.; Bao, C.; Su, S.; Gui, J. Global Analysis of Transcriptome and Translatome Revealed That Coordinated WNT and FGF Regulate the Carapacial Ridge Development of Chinese Soft-Shell Turtle. Int. J. Mol. Sci. 2021, 22, 12441. [Google Scholar] [CrossRef]
- Cebra-Thomas, J.; Tan, F.; Sistla, S.; Estes, E.; Bender, G.; Kim, C.; Riccio, P.; Gilbert, S. How the turtle forms its shell: A paracrine hypothesis of carapace formation. J. Exp. Zool. Part B 2005, 304, 558–569. [Google Scholar] [CrossRef]
- Yntema, C. A series of stages in the embryonic development of Chelydra serpentina. J. Morphol. 1968, 125, 219–251. [Google Scholar] [CrossRef]
- Tokita, M.; Kuratani, S. Normal Embryonic Stages of the Chinese Softshelled Turtle Pelodiscus sinensis (Trionychidae). Zool. Sci. 2001, 18, 705–715. [Google Scholar] [CrossRef]
- Grabherr, M.; Haas, B.; Yassour, M.; Levin, J.; Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Azaman, S.N.A.; Wong, D.C.; Tan, S.W.; Yusoff, F.M.; Nagao, N.; Yeap, S.K. De novo transcriptome analysis of Chlorella sorokiniana: Effect of glucose assimilation, and moderate light intensity. Sci. Rep. 2020, 10, 17331. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, C.; Zhu, J.; Shi, H.; Luo, J.; Zhao, W.; Shi, H.; Xu, H.; Wang, H.; Loor, J.J. Comprehensive transcriptome profiling of dairy goat mammary gland identifies genes and networks crucial for lactation and fatty acid metabolism. Front. Genet. 2020, 11, 878. [Google Scholar] [CrossRef] [PubMed]
- Gallizioli, M.; Miró-Mur, F.; Otxoa-de-Amezaga, A.; Cugota, R.; Salas-Perdomo, A.; Justicia, C.; Brait, V.H.; Ruiz-Jaén, F.; Arbaizar-Rovirosa, M.; Pedragosa, J. Dendritic cells and microglia have non-redundant functions in the inflamed brain with protective effects of type 1 cDCs. Cell Rep. 2020, 33, 108291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, P.; Liu, T.; Qiao, D.; Hu, Q.; Su, S. Identification and functional analysis of SOX transcription factors in the genome of the Chinese soft-shell turtle (Pelodiscus sinensis). Comp. Biochem. Physiol. B Mol. Biol. 2020, 242, 110407. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.; Mukherjee, S.; Ebert, B.; Gillette, M.; Paulovich, A.; Pomeroy, S.; Golub, T.; Lander, E.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Van der Auwera, G.; Carneiro, M.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, P.; Zhou, Q.; Li, X.; Ding, S.; Su, S.; Zhang, X.; Yang, X.; Zhou, W.; Wan, Q.; et al. Screening and characterisation of sex differentiation-related long non-coding RNAs in Chinese soft-shell turtle (Pelodiscus sinensis). Sci. Rep. 2018, 8, 8630. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Tipton, C.; Wolcott, R.; Sanford, N.; Miller, C.; Pathak, G.; Silzer, T.; Sun, J.; Fleming, D.; Rumbaugh, K.; Little, T.; et al. Patient genetics is linked to chronic wound microbiome composition and healing. PLoS Pathog. 2020, 16, e1008511. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordero, G.A. The turtle’s shell. Curr. Biol. 2017, 27, R168–R169. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; von Kries, J.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Nakamura, Y.; Obama, K.; Furukawa, Y. Identification of SP5 as a downstream gene of the β-catenin/Tcf pathway and its enhanced expression in human colon cancer. Int. J. Oncol. 2005, 27, 1483–1487. [Google Scholar] [CrossRef]
- Andre, P.; Song, H.; Kim, W.; Kispert, A.; Yang, Y. Wnt5a and Wnt11 regulate mammalian anterior-posterior axis elongation. Development 2015, 142, 1516–1527. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Bradley, A.; McMahon, A.; Jones, S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 1999, 126, 1211–1223. [Google Scholar] [CrossRef]
- Gao, B.; Song, H.; Bishop, K.; Elliot, G.; Garrett, L.; English, M.; Andre, P.; Robinson, J.; Sood, R.; Minami, Y.; et al. Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev. Cell. 2011, 20, 163–176. [Google Scholar] [CrossRef]
- Gros, J.; Serralbo, O.; Marcelle, C. WNT11 acts as a directional cue to organize the elongation of early muscle fibres. Nature 2009, 457, 589–593. [Google Scholar] [CrossRef]
- Cha, S.-W.; Tadjuidje, E.; Tao, Q.; Wylie, C.; Heasman, J. Wnt5a and Wnt11 interact in a maternal Dkk1-regulated fashion to activate both canonical and non-canonical signaling in Xenopus axis formation. Development 2008, 135, 3719–3729. [Google Scholar] [CrossRef]
- Jin, L.; Wu, J.; Bellusci, S.; Zhang, J.-S. Fibroblast growth factor 10 and vertebrate limb development. Front. Genet. 2019, 9, 705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohuchi, H.; Nakagawa, T.; Yamamoto, A.; Araga, A.; Ohata, T.; Ishimaru, Y.; Yoshioka, H.; Kuwana, T.; Nohno, T.; Yamasaki, M.; et al. The mesenchymal factor, FGF10, initiates and maintains the outgrowth of the chick limb bud through interaction with FGF8, an apical ectodermal factor. Development 1997, 124, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Weinstein, M.; Li, C.; Naski, M.; Cohen, R.; Ornitz, D.; Leder, P.; Deng, C. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development 1998, 125, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Capdevila, J.; Büscher, D.; Itoh, T.; Rodríguez Esteban, C.; Izpisúa Belmonte, J. WNT signals control FGF-dependent limb initiation and AER induction in the chick embryo. Cell 2001, 104, 891–900. [Google Scholar] [CrossRef]
- Gao, B.; Ajima, R.; Yang, W.; Li, C.; Song, H.; Anderson, M.; Liu, R.; Lewandoski, M.; Yamaguchi, T.; Yang, Y. Coordinated directional outgrowth and pattern formation by integration of Wnt5a and Fgf signaling in planar cell polarity. Development 2018, 145, dev163824145. [Google Scholar] [CrossRef]
- Wang, B.; Sinha, T.; Jiao, K.; Serra, R.; Wang, J. Disruption of PCP signaling causes limb morphogenesis and skeletal defects and may underlie Robinow syndrome and brachydactyly type B. Hum. Mol. Genet. 2011, 20, 271–285. [Google Scholar] [CrossRef]
- Meganathan, K.; Sotiriadou, I.; Natarajan, K.; Hescheler, J.; Sachinidis, A. Signaling molecules, transcription growth factors and other regulators revealed from in-vivo and in-vitro models for the regulation of cardiac development. Int. J. Cardiol. 2015, 183, 117–128. [Google Scholar] [CrossRef]
- Santos, J.; Mendes-Silva, L.; Afonso, V.; Martins, G.; Machado, R.; Lopes, J.; Cancela, L.; Futschik, M.; Sachinidis, A.; Gavaia, P.; et al. Exogenous WNT5A and WNT11 proteins rescue CITED2 dysfunction in mouse embryonic stem cells and zebrafish morphants. Cell Death Dis. 2019, 10, 582. [Google Scholar] [CrossRef]
- Norrie, J.; Lewandowski, J.; Bouldin, C.; Amarnath, S.; Li, Q.; Vokes, M.; Ehrlich, L.; Harfe, B.; Vokes, S. Dynamics of BMP signaling in limb bud mesenchyme and polydactyly. Dev. Biol. 2014, 393, 270–281. [Google Scholar] [CrossRef]
- Gelse, K.; Pöschl, E.; Aigner, T. Collagens—structure, function, and biosynthesis. Adv. Drug. Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef]
- Büttner, C.; Skupin, A.; Rieber, E. Transcriptional activation of the type I collagen genes COL1A1 and COL1A2 in fibroblasts by interleukin-4: Analysis of the functional collagen promoter sequences. J. Cell. Physiol. 2004, 198, 248–258. [Google Scholar] [CrossRef]
- Rossi, P.; Karsenty, G.; Roberts, A.; Roche, N.; Sporn, M.; de Crombrugghe, B. A nuclear factor 1 binding site mediates the transcriptional activation of a type I collagen promoter by transforming growth factor-β. Cell 1988, 52, 405–414. [Google Scholar] [CrossRef]
- Jobling, A.; Nguyen, M.; Gentle, A.; McBrien, N. Isoform-specific changes in scleral transforming growth factor-β expression and the regulation of collagen synthesis during myopia progression. J. Biol. Chem. 2004, 279, 18121–18126. [Google Scholar] [CrossRef] [PubMed]
- Togo, S.; Sato, T.; Sugiura, H.; Wang, X.; Basma, H.; Nelson, A.; Liu, X.; Bargar, T.W.; Sharp, J.G.; Rennard, S.I. Differentiation of embryonic stem cells into fibroblast-like cells in three-dimensional type I collagen gel cultures. Vitr. Cell Dev. An. 2011, 47, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Solar ultraviolet irradiation reduces collagen in photoaged human skin by blocking transforming growth factor-β type II receptor/Smad signaling. Am. J. Pathol. 2004, 165, 741–751. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, X.; Fu, A.; Wang, H. Extraction, identification and physicochemical properties of collagen from skirt edge of Chinese turtles. Food Ferment. Ind. 2019, 45, 176–182. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Fgf8 | CAACGGCAAAGGCAAAGACT | CGGGTGAAAGCCATGTACCA |
| Bmp5 | AGTAGAACGCAGCATAGCCC | CCATCAGGATTCTTCCAGGATG |
| Msx2 | ATTCAAGAGGCCGGGAGATA | GGGTTCTGGGCTTCCTGTTA |
| Wnt5a | CAGAGCACACTGTTCGGTGA | TCAAAGCAGGATGTGACCCAT |
| Wnt11 | GTGGCTGTGGAAAAATGAAGC | CTGATTCAGAGCAAGGACGG |
| Sp5 | TCTCCCGCTCTCTGAACACA | GGGTTGAGTGATGGGCATTC |
| Pax1 | CGCCTCTTTGAATGCAGTTGT | TTCCAGTCCCGTAGCCAAAC |
| Lef1 | GCATCCTCCAGCTCCTGATAT | CTGCCTGAATCCACTCGAGAT |
| GAPDH | CAGAGATGACGACACGCTTAG | CCATTTTCCAGGAGCGTGAT |
| Sample | Raw Reads | Clean Reads (%) | Low Quality (%) | Adapter (%) |
|---|---|---|---|---|
| TotalCrTK14 | 119,083,328 | 118,663,122 (99.65%) | 321,774 (0.27%) | 98,160 (0.09%) |
| TotalCrTK15 | 112,965,840 | 112,517,582 (99.60%) | 339,726 (0.30%) | 107,652 (0.10%) |
| TotalCrTK16 | 119,310,526 | 118,880,308 (99.64%) | 325,846 (0.27%) | 102,934 (0.09%) |
| Key Genes in M. reevesii | Key Genes in P. sinensis | ||
|---|---|---|---|
| Cyan | Wnt5a, Wnt11, Wnt-1, Wnt4, Wnt6, Wnt10b, Wnt16, Fgf5, Fgf6, Fgf20, Lef1, Mapk8 (JNK); | Black | Wnt5b, Wnt11, Wnt3, Wnt4i, Fgf7, Fgf6, Fgf19, Lef1; |
| Dark magenta | Mapk9 (JNK), Fgfr1, Wnt2b; | Pink | JNK, Fgfr2, Wnt16; |
| Coral 3 | Gremlin1; | Coral1 | Col1a, Itga7; |
| Navajo white 3 | Shh, Ptch1, Ptch2, Col1a1, Col1a2, Tgfb2, Itga2, Itga5, Itga7, Itga10, Itga11. | Dark green | Col1a1, Col1a2, Col24a1, Col5a2, Itga8, Itga11, ItgaV, Smad6, Smad4, Tgfb2, Bmp6, Wnt5a, Dkk2. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Xia, Y.; Li, S.; Chen, T.; Zhang, J.; Weng, Z.; Zheng, H.; Jin, M.; Bao, C.; Su, S.; et al. Gene Regulation during Carapacial Ridge Development of Mauremys reevesii: The Development of Carapacial Ridge, Ribs and Scutes. Genes 2022, 13, 1676. https://doi.org/10.3390/genes13091676
Yang J, Xia Y, Li S, Chen T, Zhang J, Weng Z, Zheng H, Jin M, Bao C, Su S, et al. Gene Regulation during Carapacial Ridge Development of Mauremys reevesii: The Development of Carapacial Ridge, Ribs and Scutes. Genes. 2022; 13(9):1676. https://doi.org/10.3390/genes13091676
Chicago/Turabian StyleYang, Jiayu, Yingying Xia, Shaohu Li, Tingting Chen, Jilong Zhang, Zhiyuan Weng, Huiwei Zheng, Minxuan Jin, Chuanhe Bao, Shiping Su, and et al. 2022. "Gene Regulation during Carapacial Ridge Development of Mauremys reevesii: The Development of Carapacial Ridge, Ribs and Scutes" Genes 13, no. 9: 1676. https://doi.org/10.3390/genes13091676
APA StyleYang, J., Xia, Y., Li, S., Chen, T., Zhang, J., Weng, Z., Zheng, H., Jin, M., Bao, C., Su, S., Liang, Y., & Zhang, J. (2022). Gene Regulation during Carapacial Ridge Development of Mauremys reevesii: The Development of Carapacial Ridge, Ribs and Scutes. Genes, 13(9), 1676. https://doi.org/10.3390/genes13091676