
Improving the Utilization of STRmix™ Variance Parameters as Semi-Quantitative Profile Modeling Metrics


Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction, Amplification, Capillary Electrophoresis, and Analysis of Unchallenged DNA Samples
2.2. Construction, Amplification, CE, and Analysis of Challenged DNA Samples

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Number of Samples | Input Amounts | Replicate Amplifications | Replicate STRmix Interpretations |
|---|---|---|---|---|
| Single-source, nominal-input | 14 | 2 ng | 1 | 10 |
| Single-source dilution series (higher level) | 2 | 8 ng, 4 ng, 2 ng, 1 ng, 500 pg, 250 pg | 2 | 1 |
| Single-source dilution series (lower level) | 2 | 125 pg, 63 pg | 4 | 1 |
| Donor Number (Donor Set) | Mixture Ratio | Input Amounts | Replicate Amplifications |
|---|---|---|---|
| 2-person (set 1) | 9:1 | 2 ng, 1 ng, 870 pg, 750 pg, 500 pg, 380 pg, 250 pg, 125 pg, 63 pg | 2 |
| 2-person (set 1) | 49:1 | 2.5 ng, 1.9 ng, 1.25 ng, 625 pg, 313 pg | 2 |
| 2-person (set 1) | 99:1 | 2.5 ng, 1.25 ng, 625 pg | 2 |
| 2-person (set 2) | 1:1 | 800 pg, 400 pg, 200 pg, 100 pg, 50 pg, 25 pg | 1 |
| 2-person (set 2) | 3:1 | 800 pg, 400 pg, 348 pg, 300 pg, 200 pg, 152 pg, 100 pg, 50 pg, 25 pg | 1 |
| 3-person (set 1) | 3:2:1 | 1.2 ng, 600 pg, 522 pg, 450 pg, 300 pg, 228 pg, 150 pg, 75 pg, 38 pg | 2 |
| 3-person (set 1) | 10:5:1 | 3.2 ng, 1.6 ng, 1.4 ng, 1.2 ng, 800 pg, 608 pg, 400 pg, 200 pg, 100 pg | 2 |
| 3-person (set 1) | 100:100:4 | 1.28 ng, 625 pg, 325 pg | 2 |
| 3-person (set 2) | 1:1:1 | 1.2 ng, 600 pg, 300 pg, 150 pg, 75 pg, 38 pg | 1 |
| 3-person (set 2) | 3:2:1 | 1.2 ng, 522 pg, 300 pg, 150 pg, 38 pg | 2 |
| 3-person (set 2) | 10:5:1 | 3.2 ng, 1.4 ng, 800 pg, 400 pg, 100 pg | 2 |
| 3-person (set 2) | 100:100:4 | 1.28 ng, 638 pg, 319 pg | 2 |
| 4-person (set 1) | 4:3:2:1 | 2 ng, 1 ng, 870 pg, 750 pg, 500 pg, 380 pg, 250 pg, 125 pg, 63 pg | 2 |
| 4-person (set 1) | 10:5:2:1 | 3.6 ng, 1.8 ng, 1.6 ng, 1.4 ng, 900 pg, 684 pg, 450 pg, 225 pg, 113 pg | 2 |
| 4-person (set 1) | 100:100:100:6 | 1.28 ng, 625 pg, 325 pg | 2 |
| 4-person (set 2) | 1:1:1:1 | 1.6 ng, 800 pg, 400 pg, 200 pg, 100 pg, 50 pg | 1 |
| 4-person (set 2) | 4:3:2:1 | 2 ng, 870 pg, 500 pg, 250 pg, 63 pg | 2 |
| 4-person (set 2) | 10:5:2:1 | 3.6 ng, 1.6 ng, 900 pg, 450 pg, 113 pg | 2 |
| 4-person (set 2) | 100:100:100:6 | 1.28 ng, 638 pg, 319 pg | 2 |
| Donor Number | Mixture Ratio | Treatment |
|---|---|---|
| 2-person | 3:1 | Hematin: 400 µM, 475 µM, 550 µM, 625 µM, 700 µM |
| 10:1 | Hematin: 400 µM, 475 µM, 550 µM, 625 µM, 700 µM | |
| 3:1 | Humic acid: 200 ng/µL, 250 ng/µL, 300 ng/µL, 350 ng/µL, 400 ng/µL | |
| 10:1 | Humic acid: 200 ng/µL, 250 ng/µL, 300 ng/µL, 350 ng/µL, 400 ng/µL | |
| 3-person | 3:2:1 | Hematin: 400 µM, 475 µM, 550 µM, 625 µM, 700 µM |
| 10:5:1 | Hematin: 400 µM, 475 µM, 550 µM, 625 µM, 700 µM | |
| 3:2:1 | Humic acid: 200 ng/µL, 250 ng/µL, 300 ng/µL, 350 ng/µL, 400 ng/µL | |
| 10:5:1 | Humic acid: 200 ng/µL, 250 ng/µL, 300 ng/µL, 350 ng/µL, 400 ng/µL | |
| 4-person | 4:3:2:1 | Hematin: 400 µM, 475 µM, 550 µM, 625 µM, 700 µM |
| 10:5:2:1 | Hematin: 400 µM, 475 µM, 550 µM, 625 µM, 700 µM | |
| 4:3:2:1 | Humic acid: 200 ng/µL, 250 ng/µL, 300 ng/µL, 350 ng/µL, 400 ng/µL | |
| 10:5:2:1 | Humic acid: 200 ng/µL, 250 ng/µL, 300 ng/µL, 350 ng/µL, 400 ng/µL |
| Dry Heat Treatment Number | Dry Heat Exposure Time |
|---|---|
| 1 | 5.75 h |
| 2 | 12.13 h |
| 3 | 19.42 h |
| 4 | 27.73 h |
| 5 | 37.32 h |
| 6 | 48.50 h |
| 7 | 61.70 h |
| 8 | 77.52 h |
| 9 | 96.83 h |
| 10 | 120.93 h |
| 11 | 151.85 h |
| 12 | 192.97 h |
| 13 | 250.32 h |
| 14 | 335.88 h |
| Donor Number | Mixture Ratio | C1 Dry Heat Treatments | C2 Dry Heat Treatments | C3 Dry Heat Treatments | C4 Dry Heat Treatments |
|---|---|---|---|---|---|
| Single source #1 | - | 1,3,4,5,6,7,9,13,10 | - | - | - |
| Single source #2 | - | 1,2,3,4,5,6,8,10,14 | - | - | - |
| Single source #3 | - | 1,2,4,5,6,9,10,11,12 | - | - | - |
| Single source #4 | - | 1,2,4,5,8,9,11,12,13 | - | - | - |
| 2-person | 3:1 | 1,3,4,5,6,7,9,13,10 | 1,2,3,4,5,6,8,10,14 | - | - |
| 10:1 | 1,3,4,5,6,7,9,13,10 | 1,2,3,4,5,6,8,10,14 | - | - | |
| 3-person | 3:2:1 | 1,3,4,5,6,7,9,13,10 | 1,2,3,4,5,6,8,10,14 | 1,2,4,5,6,9,10,11,12 | - |
| 10:5:1 | 1,3,4,5,6,7,9,13,10 | 1,2,3,4,5,6,8,10,14 | 1,2,4,5,6,9,10,11,12 | - | |
| 4-person | 4:3:2:1 | 1,3,4,5,6,7,9,13,10 | 1,2,3,4,5,6,8,10,14 | 1,2,4,5,6,9,10,11,12 | 1,2,4,5,8,9,11,12,13 |
| 10:5:2:1 | 1,3,4,5,6,7,9,13,10 | 1,2,3,4,5,6,8,10,14 | 1,2,4,5,6,9,10,11,12 | 1,2,4,5,8,9,11,12,13 |
| Donor Number (Donor Set) | Mixture Ratio | Input Amounts (Total DNA) |
|---|---|---|
| 2-person (set 1) | 9:1 | 28 ng |
| 2-person (set 1) | 99:1 | 25.5 ng |
| 2-person (set 2) | 1:1 | 29.3 ng |
| 2-person (set 2) | 3:1 | 20.9 ng |
| 3-person (set 1) | 3:2:1 | 24 ng |
| 3-person (set 1) | 10:5:1 | 29 ng |
| 3-person (set 1) | 100:100:4 | 28.5 ng |
| 3-person (set 2) | 1:1:1 | 20.4 ng |
| 3-person (set 2) | 3:2:1 | 17.8 ng |
| 3-person (set 2) | 10:5:1 | 12.7 ng |
| 3-person (set 2) | 100:100:4 | 17.1 ng |
| 4-person (set 1) | 4:3:2:1 | 32.5 ng |
| 4-person (set 1) | 10:5:2:1 | 29.4 ng |
| 4-person (set 1) | 100:100:100:6 | 30 ng |
| 4-person (set 2) | 1:1:1:1 | 20.3 ng |
| 4-person (set 2) | 4:3:2:1 | 15.4 ng |
| 4-person (set 2) | 10:5:2:1 | 11.9 ng |
| 4-person (set 2) | 100:100:100:6 | 22.0 ng |
| Ground Truth Donor Number (Donor Set) | STRmix Donor Number Setting (NOC-1 or NOC-2) | Mixture Ratio | Input Amounts (Total DNA) |
|---|---|---|---|
| 2-person (set 1) | 1 | 49:1 | 313 pg |
| 2-person (set 1) | 1 | 99:1 | 2.5 ng, 1.25 ng |
| 3-person (set 1) | 2 | 3:2:1 | 1.2 ng, 600 pg, 522 pg, 450 pg, 300 pg, 228 pg, 150 pg, 75 pg, 38 pg |
| 3-person (set 2) | 2 | 3:2:1 | 38 pg |
| 3-person (set 1) | 2 | 10:5:1 | 800 pg, 608 pg, 400 pg, 200 pg, 100 pg |
| 3-person (set 2) | 2 | 10:5:1 | 100 pg |
| 3-person (set 1) | 2 | 100:100:4 | 1.28 ng, 625 pg, 325 pg |
| 3-person (set 2) | 2 | 100:100:4 | 1.28 ng, 625 pg, 325 pg |
| 4-person (set 1) | 2 | 4:3:2:1 | 125 pg, 63 pg |
| 4-person (set 1) | 2 | 10:5:2:1 | 113 pg |
| 4-person (set 2) | 2 | 10:5:2:1 | 113 pg |
| 4-person (set 1) | 3 | 4:3:2:1 | 2 ng, 1 ng, 870 pg, 750 pg, 500 pg, 380 pg, 250 pg |
| 4-person (set 2) | 3 | 4:3:2:1 | 63 pg |
| 4-person (set 1) | 3 | 10:5:2:1 | 3.6 ng, 1.8 ng, 1.6 ng, 1.4 ng, 684 pg, 450 pg, 225 pg |
| 4-person (set 2) | 3 | 10:5:2:1 | 450 pg |
| 4-person (set 1) | 3 | 100:100:100:6 | 1.28 ng, 638 ng, 319 ng |
| 4-person (set 2) | 3 | 100:100:100:6 | 1.28 ng, 625 ng, 325 ng |
| Donor Number | Mixture Ratio | Input Amounts | Replicate Amps |
|---|---|---|---|
| 2-person (CEPH 1347-02, HL60) | 9:1 | 2 ng, 1 ng, 870 pg, 750 pg, 500 pg, 380 pg, 250 pg, 125 pg, 63 pg | 2 |
| 49:1 | 2.5 ng, 1.9 ng, 1.25 ng, 625 pg, 313 pg | 2 | |
| 99:1 | 2.5 ng, 1.25 ng, 625 pg | 2 | |
| 1:1 | 800 pg, 400 pg, 200 pg, 100 pg, 50 pg, 25 pg | 1 | |
| 3:1 | 800 pg, 400 pg, 348 pg, 300 pg, 200 pg, 152 pg, 100 pg, 50 pg, 25 pg | 1 | |
| 3-person (2800 M, HL60, CEPH 1347-02) | 3:2:1 | 1.2 ng, 600 pg, 522 pg, 450 pg, 300 pg, 228 pg, 150 pg, 75 pg, 38 pg | 2 |
| 10:5:1 | 3.2 ng, 1.6 ng, 1.4 ng, 1.2 ng, 800 pg, 608 pg, 400 pg, 200 pg, 100 pg | 2 | |
| 100:100:4 | 1.28 ng, 625 pg, 325 pg | 2 | |
| 1:1:1 | 1.2 ng, 600 pg, 300 pg, 150 pg, 75 pg, 38 pg | 1 | |
| 3:2:1 | 1.2 ng, 522 pg, 300 pg, 150 pg, 38 pg | 2 | |
| 10:5:1 | 3.2 ng, 1.4 ng, 800 pg, 400 pg, 100 pg | 2 | |
| 100:100:4 | 1.28 ng, 638 pg, 319 pg | 2 |
3. Results
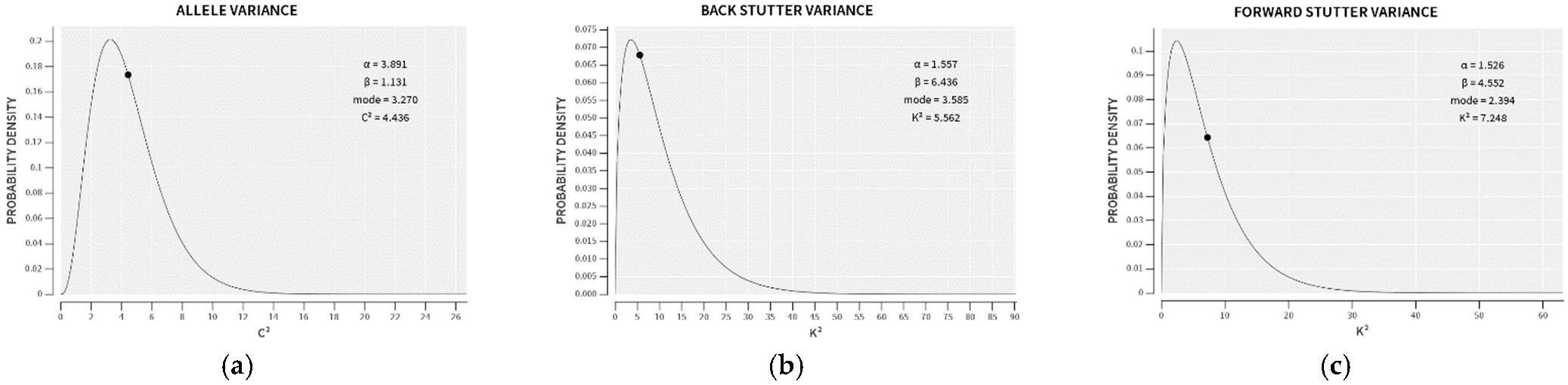
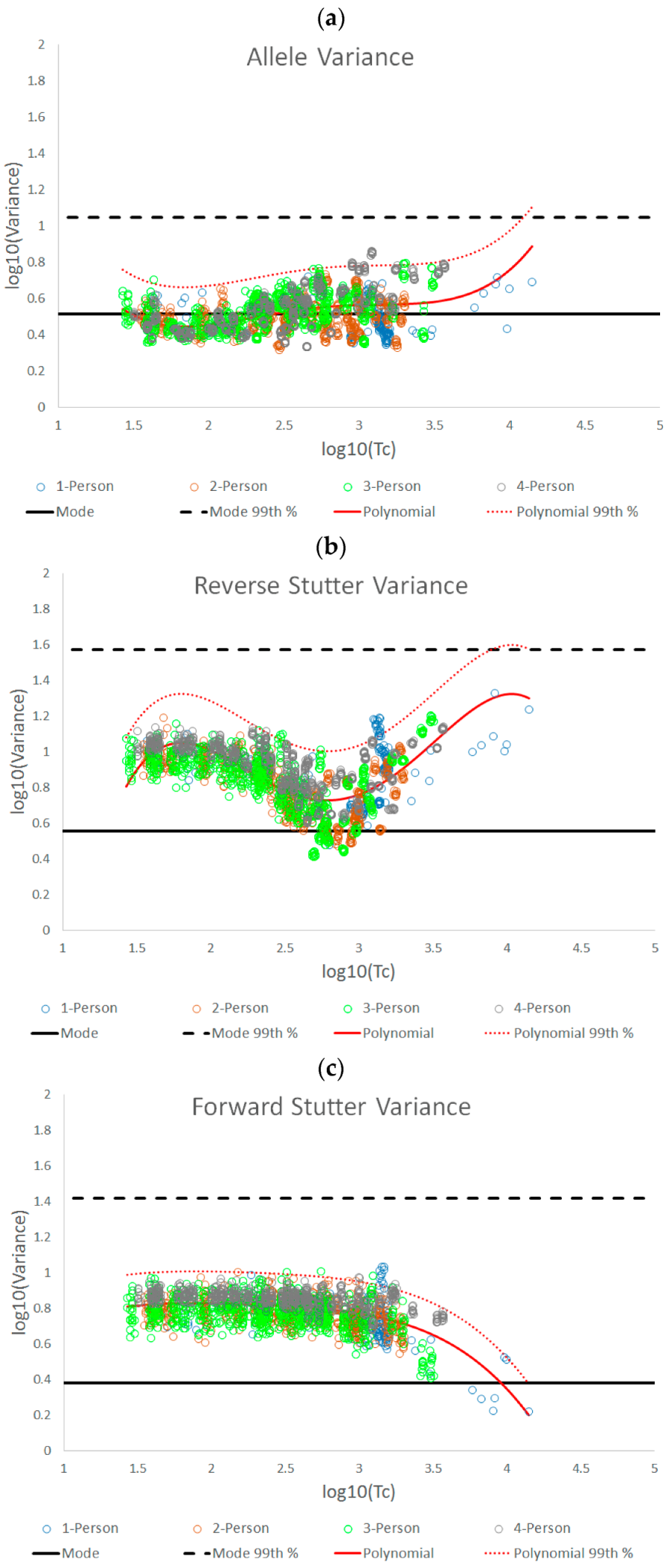
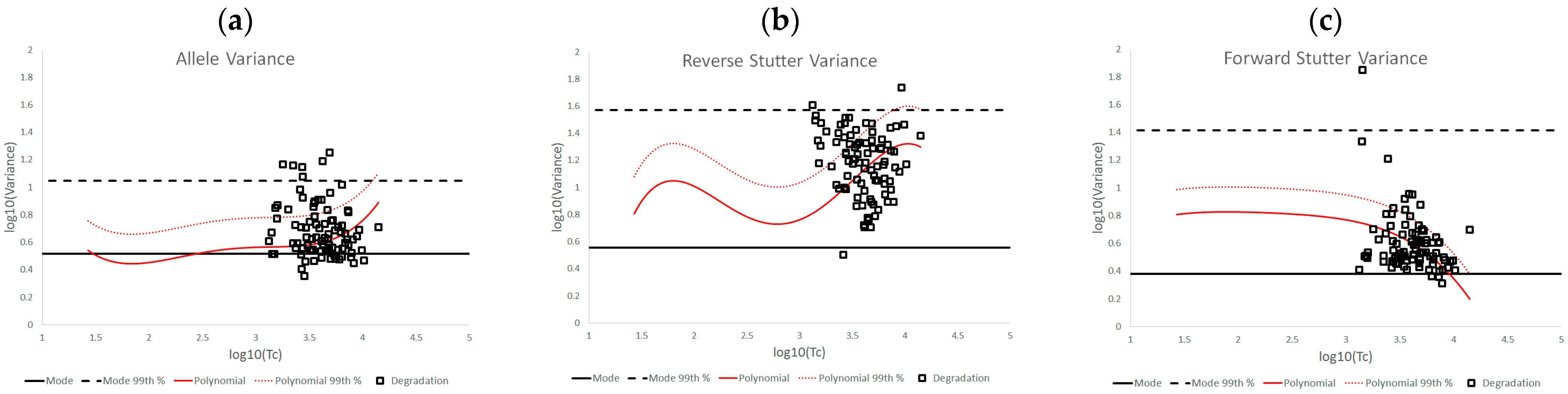
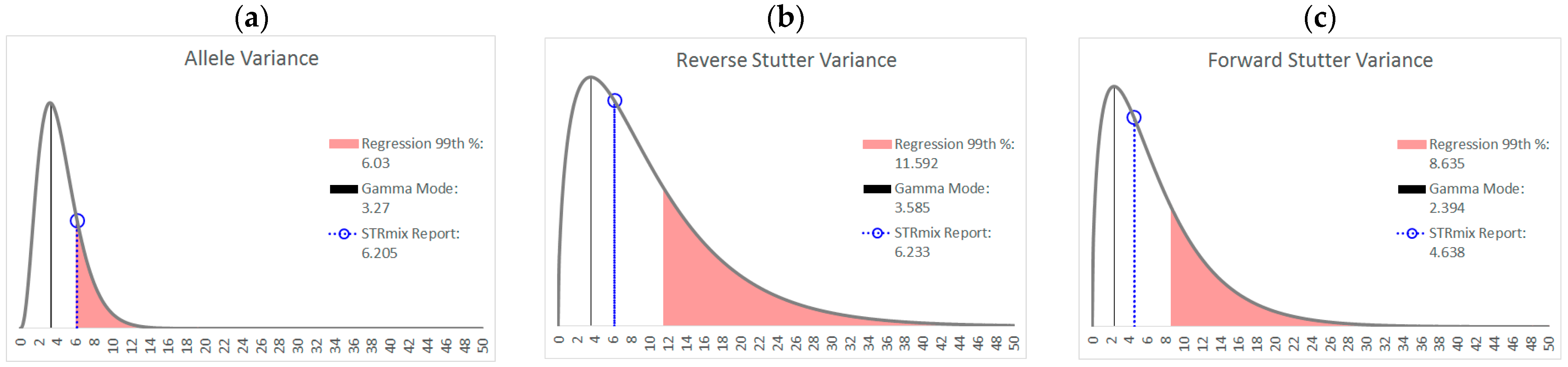
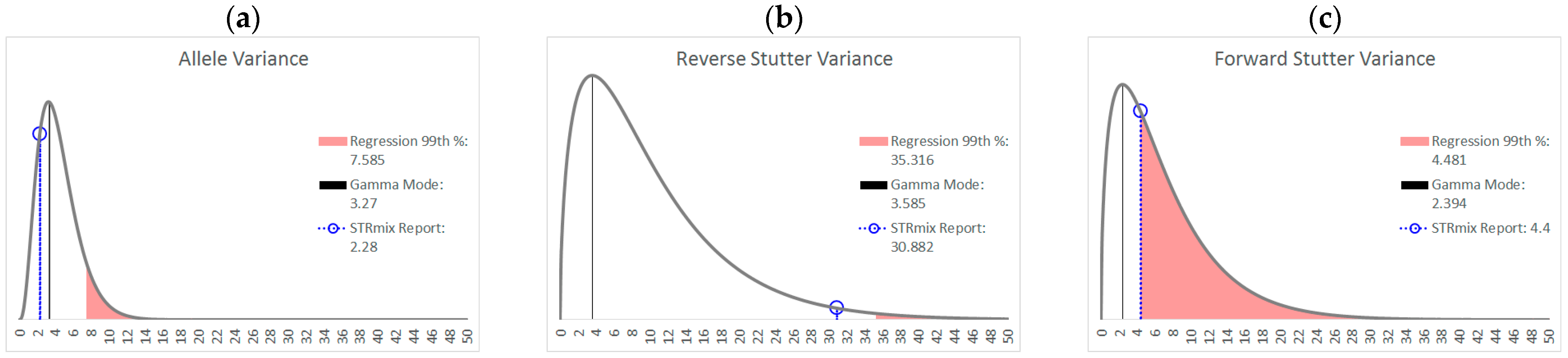
3.1. Trends in Allele and Stutter Variances with Increasing Peak Height
| Allele | Reverse Stutter | Forward Stutter | |
|---|---|---|---|
| Polynomial regression formula | y = 0.1122x4 − 1.2206x3 + 4.8535x2 − 8.2618x + 5.5312 | y = −0.2892x4 + 3.3258x3 − 13.619x2 + 23.456x − 13.404 | y = −0.0348x4 + 0.313x3 − 1.0891x2 + 1.7016x − 0.1678 |
| Jarque-Bera test for normality of the residuals | p = 0.1503 | p = 0.2395 | p = 0.0275 |
| 99th Percentile (+2.326 SD) | +0.2156 | +0.2755 | +0.1779 |
| Allele | Reverse Stutter | Forward Stutter | |
|---|---|---|---|
| α | 3.891 | 1.557 | 1.526 |
| β | 1.131 | 6.436 | 4.552 |
| Mode | 3.270 | 3.585 | 2.394 |
| 99th Percentile | 11.16 | 37.24 | 26.06 |
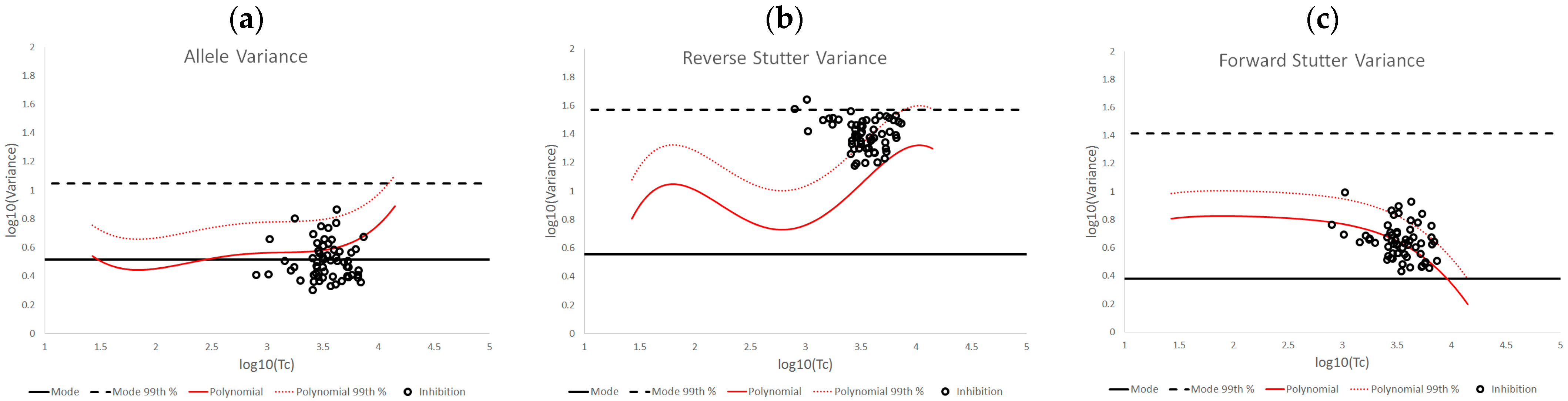
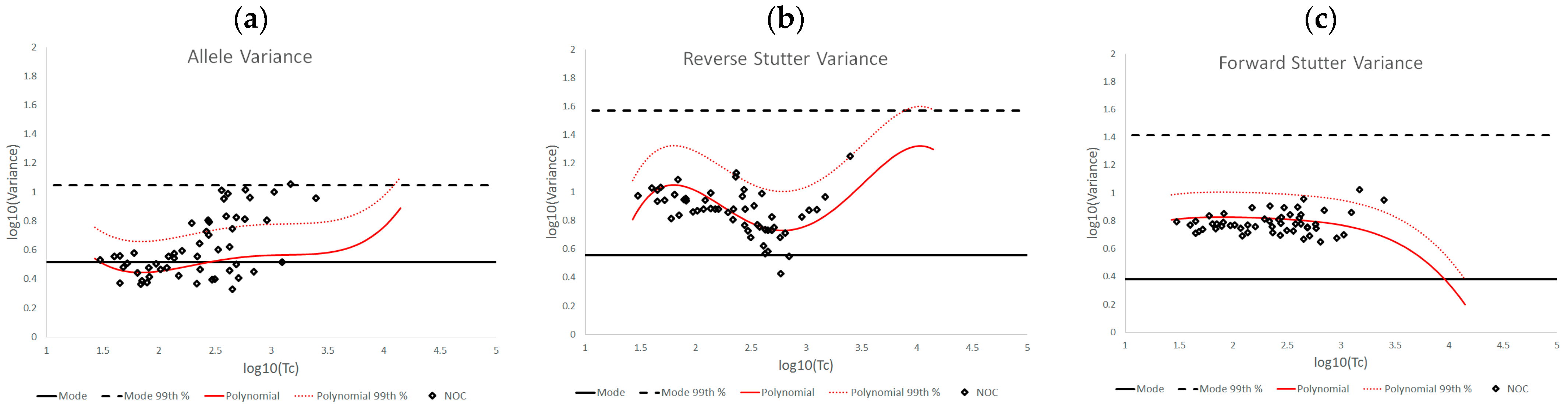
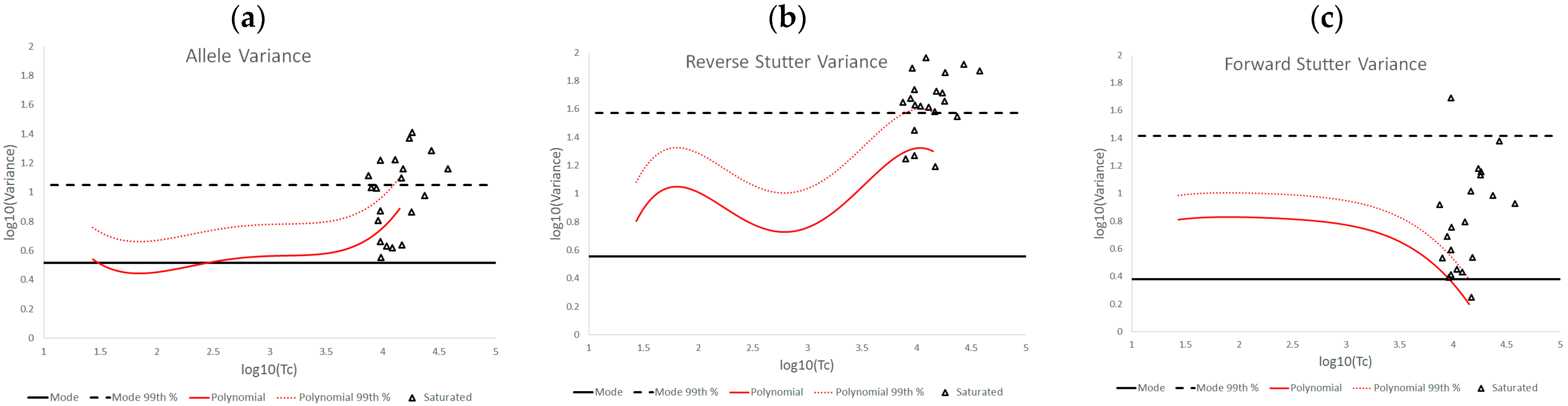
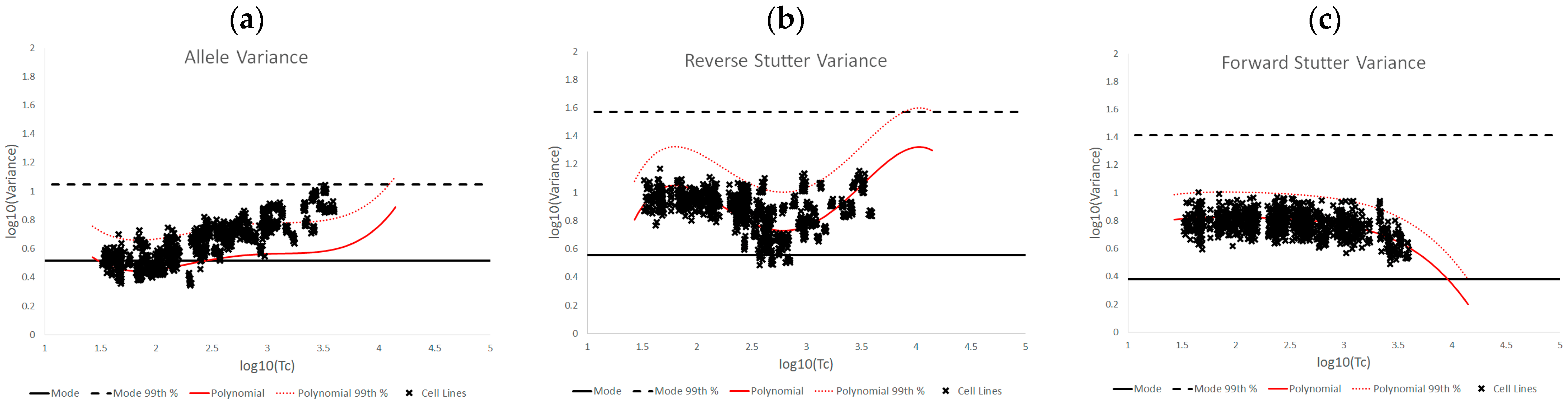
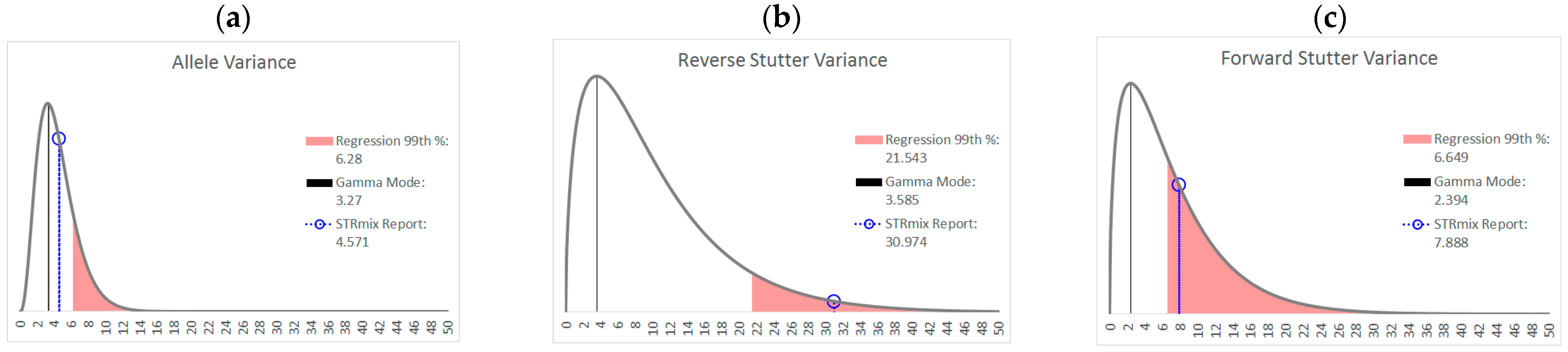
3.2. Trends in Allele and Stutter Variance under Challenging Amplification/Interpretation Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clayton, T.M.; Whitaker, J.P.; Sparkes, R.; Gill, P. Analysis and interpretation of mixed forensic stains using DNA STR profiling. Forensic. Sci. Int. 1998, 91, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Bright, J.A.; Buckleton, J. The interpretation of single source and mixed DNA profiles. Forensic. Sci. Int. Genet. 2013, 7, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Buckleton, J.; Bright, J.A. Factors affecting peak height variability for short tandem repeat data. Forensic. Sci. Int. Genet. 2016, 21, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Bille, T.W.; Weitz, S.M.; Coble, M.D.; Buckleton, J.; Bright, J.A. Comparison of the performance of different models for the interpretation of low level mixed DNA profiles. Electrophoresis 2014, 35, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Bieber, F.R.; Buckleton, J.; Budowle, B.; Butler, J.M.; Coble, M.D. Evaluation of forensic DNA mixture evidence: Protocol for evaluation, interpretation, and statistical calculations using the combined probability of inclusion. BMC Genet. 2016, 17, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckleton, J.; Bright, J.A.; Gittelson, S.; Moretti, T.; Onorato, A.J.; Bieber, F.R.; Budowle, B.; Taylor, D. The Probabilistic Genotyping Software STRmix: Utility and Evidence for its Validity. J. Forensic. Sci. 2019, 64, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Bleka, Ø.; Storvik, G.; Gill, P. EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts. Forensic. Sci. Int. Genet. 2016, 21, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Bright, J.A.; Taylor, D.; Curran, J.; Buckleton, J. Developing allelic and stutter peak height models for a continuous method of DNA interpretation. Forensic. Sci. Int. Genet. 2013, 7, 296–304. [Google Scholar] [CrossRef] [PubMed]
- STRmix v2.8 User’s Manual (September 2020); Institute of Environmental Science and Research Limited: Wellington, New Zealand, 2020.
- Russell, L.; Cooper, S.; Wivell, R.; Kerr, Z.; Taylor, D.; Buckleton, J.; Bright, J. A guide to results and diagnostics within a STRmix report. WIREs Forensic. Sci. 2019, 1, e1354. [Google Scholar] [CrossRef]
- Butler, J.M.; Iyer, H.; Press, R.; Taylor, M.K.; Vallone, P.M.; Willis, S. DNA Mixture INTERPRETATION: A NIST Scientific Foundation Review. NISTIR 8351-DRAFT; 2021. Available online: https://nvlpubs.nist.gov/nistpubs/ir/2021/NIST.IR.8351-draft.pdf (accessed on 17 November 2022).
- Duke, K.; Cuenca, D.; Myers, S.; Wallin, J. Compound and conditioned likelihood ratio behavior within a probabilistic genotyping context. Genes 2022, 13, 2031. [Google Scholar] [CrossRef] [PubMed]
- How to Perform a Normality Test in Excel (Step-by-Step). Available online: https://www.statology.org/normality-test-excel/ (accessed on 12 December 2022).
- Duke, K.; Myers, P. Systematic evaluation of STRmix™ performance on degraded DNA profile data. Forensic. Sci. Int. Genet. 2020, 44, 102174. [Google Scholar] [CrossRef] [PubMed]










| % Greater Than… | Unchallenged | Inhibited | NOC −1 or −2 | Degraded | Saturated | Cell Lines | |
|---|---|---|---|---|---|---|---|
| Allele Variance | Polynomial Regression | 48.90% | 21.67% | 67.92% | 53.33% | 55.00% | 90.50% |
| 99th Percentile | 1.05% | 3.33% | 28.30% | 21.11% | 40.00% | 21.84% | |
| Mode | 47.09% | 41.67% | 58.49% | 84.44% | 100.00% | 80.38% | |
| 99th Percentile | 0.00% | 0.00% | 1.89% | 6.67% | 45.00% | 0.00% | |
| Reverse Stutter Variance | Polynomial Regression | 50.91% | 100.00% | 39.62% | 57.78% | 85.00% | 45.60% |
| 99th Percentile | 1.15% | 56.67% | 3.77% | 23.33% | 80.00% | 1.92% | |
| Mode | 95.28% | 100.00% | 96.23% | 98.89% | 100.00% | 97.37% | |
| 99th Percentile | 0.00% | 3.33% | 0.00% | 2.22% | 75.00% | 0.00% | |
| Forward Stutter Variance | Polynomial Regression | 53.15% | 48.33% | 28.30% | 44.44% | 100.00% | 37.71% |
| 99th Percentile | 1.00% | 16.67% | 3.77% | 12.22% | 70.00% | 0.51% | |
| Mode | 99.76% | 100.00% | 100.00% | 96.67% | 95.00% | 100.00% | |
| 99th Percentile | 0.00% | 0.00% | 0.00% | 1.11% | 5.00% | 0.00% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duke, K.; Myers, S.; Cuenca, D.; Wallin, J. Improving the Utilization of STRmix™ Variance Parameters as Semi-Quantitative Profile Modeling Metrics. Genes 2023, 14, 102. https://doi.org/10.3390/genes14010102
Duke K, Myers S, Cuenca D, Wallin J. Improving the Utilization of STRmix™ Variance Parameters as Semi-Quantitative Profile Modeling Metrics. Genes. 2023; 14(1):102. https://doi.org/10.3390/genes14010102
Chicago/Turabian StyleDuke, Kyle, Steven Myers, Daniela Cuenca, and Jeanette Wallin. 2023. "Improving the Utilization of STRmix™ Variance Parameters as Semi-Quantitative Profile Modeling Metrics" Genes 14, no. 1: 102. https://doi.org/10.3390/genes14010102
APA StyleDuke, K., Myers, S., Cuenca, D., & Wallin, J. (2023). Improving the Utilization of STRmix™ Variance Parameters as Semi-Quantitative Profile Modeling Metrics. Genes, 14(1), 102. https://doi.org/10.3390/genes14010102