
What Have We Learned from Patients Who Have Arboleda-Tham Syndrome Due to a De Novo KAT6A Pathogenic Variant with Impaired Histone Acetyltransferase Function? A Precise Clinical Description May Be Critical for Genetic Testing Approach and Final Diagnosis

 ,
,  ,
,  ,
,  , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Clinical History
2.3. Clinical Exome Sequencing (CES) as Previously Reported [11]
2.4. SNP Array Analysis
2.5. Methylation Studies
3. Results
3.1. Genetic Findings
3.2. 3D Modelling
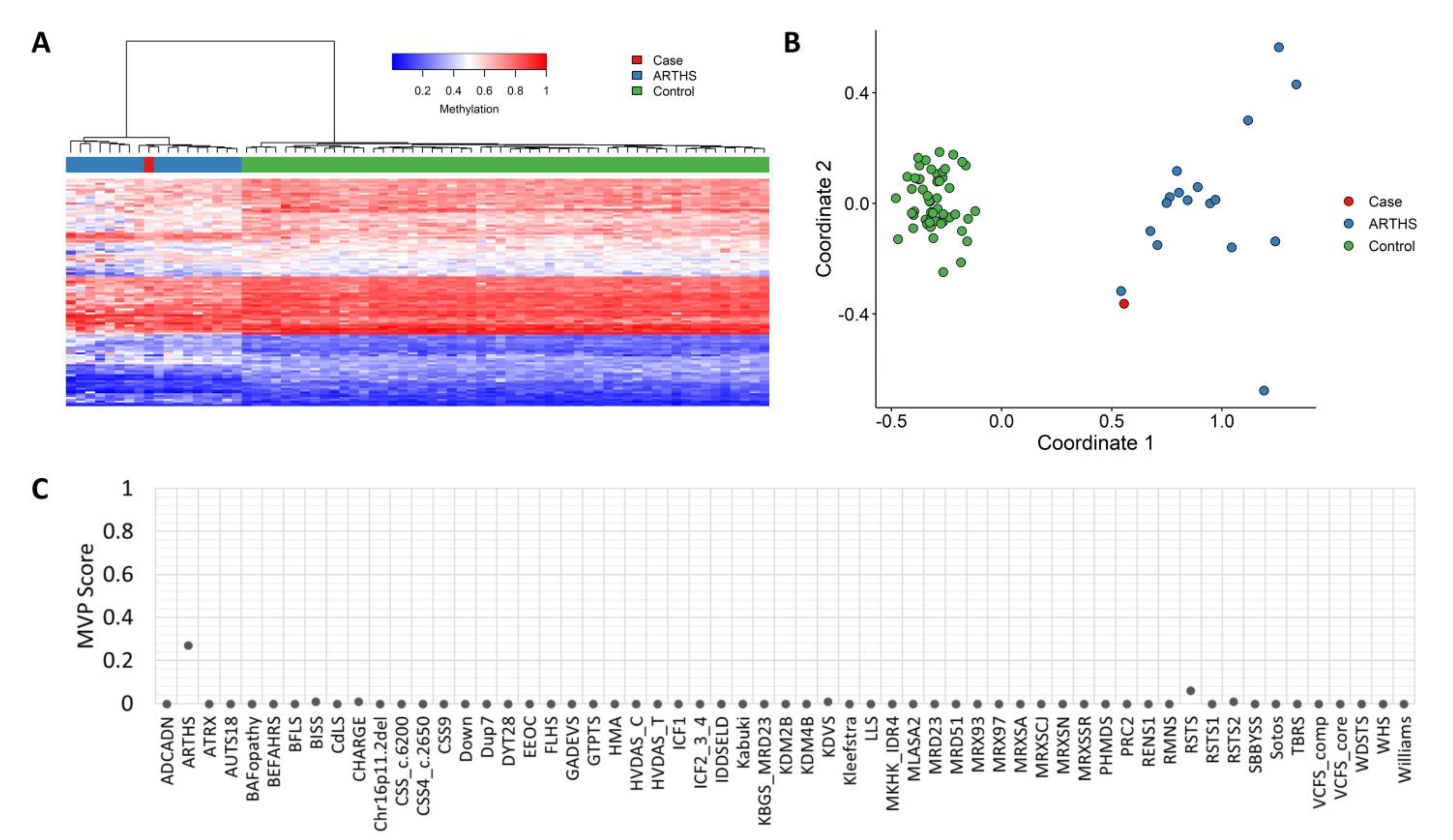
3.3. Methylation Studies
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Voss, A.K.; Collin, C.; Dixon, M.P.; Thomas, T. MOZ and retinoic acid coordinately regulate H3K9 acetylation, HOX gene expression, and segment identity. Dev. Cell 2009, 17, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef]
- Arboleda, V.A.; Lee, H.; Dorrani, N.; Zadeh, N.; Willis, M.; MacMurdo, C.F.; Manning, M.A.; Kwan, A.; Hudgins, L.; Barthelemy, F.; et al. De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. Am. J. Hum. Genet. 2015, 96, 498–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tham, E.; Lindstrand, A.; Santanim, A.; Malmgren, H.; Nesbitt, A.; Dubbs, H.A.; Zackai, E.H.; Parker, M.J.; Millan, F.; Rosenbaum, K.; et al. Dominant mutations in KAT6Acause intellectual disability with recognizable syndromic features. Am. J. Hum. Genet. 2015, 96, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genomewide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millan, F.; Cho, M.T.; Retterer, K.; Monaghan, K.G.; Bai, R.; Vitazka, P.; Everman, D.B.; Smith, B.; Angle, B.; Roberts, V.; et al. Whole exome sequencing reveals de novo pathogenic variants in KAT6A as a cause of a neurodevelopmental disorder. Am. J. Med. Genet. A 2016, 170, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novomutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Clayton-Smith, J.; O’Sullivan, J.; Daly, S.; Bhaskar, S.; Day, R.; Anderson, B.; Voss, A.K.; Thomas, T.; Biesecker, L.G.; Smith, P.; et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 2011, 89, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Campeau, P.M.; Lu, J.T.; Dawson, B.C.; Fokkema, I.F.A.C.; Robertson, S.P.; Gibbs, R.A.; Lee, B.H. The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum. Mutat. 2012, 3, 1520–1525. [Google Scholar] [CrossRef] [Green Version]
- Szakszon, K.; Salpietro, C.; Kakar, N.; Knegt, A.C.O.; Dallapiccola, B.; Borck, G. De novo mutations of the gene encoding the histone acetyltransferase KAT6B in two patients with Say- Barber/Biesecker/Young-Simpson syndrome. Am. J. Med. Genet. A 2013, 161, 884–888. [Google Scholar] [CrossRef]
- Laforgia, N.; De Cosmo, L.; Palumbo, O.; Ranieri, C.; Sesta, M.; Capodiferro, D.; Pantaleo, A.; Iapicca, P.; Lastella, P.; Capozza, M.; et al. The First Case of Congenital Myasthenic Syndrome Caused by a Large Homozygous Deletion in the C-Terminal Region of COLQ (Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase) Protein. Genes 2020, 11, 1519. [Google Scholar] [CrossRef]
- Palumbo, O.; Palumbo, P.; Di Muro, E.; Cinque, L.; Petracca, A.; Carella, M.; Castori, M.A. Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features. Genes 2020, 11, 707. [Google Scholar] [CrossRef] [PubMed]
- Aref-Eshghi, E.; Bend, E.G.; Colaiacovo, S.; Caudle, M.; Chakrabarti, R.; Napier, M.; Brick, L.; Brady, L.; Carere, D.A.; Levy, M.A.; et al. Diagnostic Utility of Genome-wide DNA Methylation Testing in Genetically Unsolved Individuals with Suspected Hereditary Conditions. Am. J. Hum. Genet. 2019, 104, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Groupe DI France,; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef]
- Levy, M.A.; McConkey, H.; Kerkhof, J.; Barat-Houari, M.; Bargiacchi, S.; Biamino, E.; Bralo, M.P.; Cappuccio, G.; Ciolfi, A.; Clarke, A.; et al. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. HGG Adv. 2021, 3, 100075. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Levy, M.A.; Kerkhof, J.; Aref-Eshghi, E.; Schenkel, L.; Stuart, A.; McConkey, H.; Henneman, P.; Venema, A.; Schwartz, C.E.; et al. Clinical epigenomics: Genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet. Med. 2021, 23, 1065–1074. [Google Scholar] [CrossRef]
- Kennedy, J.; Goudie, D.; Blair, E.; Chandler, K.; Joss, S.; McKay, V.; Green, A.; Armstrong, R.; Lees, M.; Kamien, B.; et al. KAT6A Syndrome: Genotype–phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. 2019, 21, 850–860. [Google Scholar] [CrossRef]
- Lonardo, F.; Lonardo, M.S.; Acquaviva, F.; Della Monica, M.; Scarano, F.; Scarano, G. Say-Barber-Biesecker-Young-Simpson syndrome and Genitopatellar syndrome: Lumping or splitting? Clin. Genet. 2017, 95, 253–261. [Google Scholar] [CrossRef]
- Köhler, S.; Marcel, S.; Krawitz, P.; Bauer, S.; Dölken, S.; Ott, C.; Mundlos, C.; Horn, D.; Mundlos, S.; Robinson, P. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet. 2009, 85, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Köhler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.-P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019, 47, 1018–1027. [Google Scholar] [CrossRef]
- Yang, X.J. MOZ and MORF acetyltransferases: Molecular interaction, animal development and human disease. Biochim. Biophys. Acta 2015, 1853, 1818–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattioli, F.; Schaefer, E.; Magee, A.; Mark, P.; Mancini, G.M.; Dieterich, K.; Von Allmen, G.; Alders, M.; Coutton, C.; Van Slegtenhorst, M.; et al. Mutations in histone acetylase modifier BRPF1 cause an autosomal-dominant form of intellectual disability with associated ptosis. Am. J. Hum. Genet. 2017, 100, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, K.; Rousseau, J.; Littlejohn, R.O.; Kiss, C.; Lehman, A.; Rosenfeld, J.A.; Stumpel, C.T.; Stegmann, A.P.; Robak, L.; Scaglia, F.; et al. Mutations in the chromatin regulator gene BRPF1 cause syndromic intellectual disability and deficient histone acetylation. Am. J. Hum. Genet. 2017, 100, 91–104. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnostic Criterion * |
|---|
| Patent ductus arteriosus HP:0001643 |
| Abnormal facial shape HP:0001999 |
| Intellectual disability, severe HP:0010864 |
| Clinical signs and symptoms |
| Very frequent |
| Abnormal facial shape HP:0001999 |
| Broad nasal tip HP:0000455 |
| Global developmental delay HP:0001263 |
| Intellectual disability, severe HP:0010864 |
| Microcephaly HP:0000252 |
| Narrow forehead HP:0000341 |
| Neonatal hypotonia HP:0001319 |
| Poor speech HP:0002465 |
| Prominent nasal bridge HP:0000426 |
| Thin upper lip vermilion HP:0000219 |
| Frequent |
| Atrial septal defect HP:0001631 |
| Cerebral visual impairment HP:0100704 |
| Craniosynostosis HP:0001363 |
| Downturned corners of mouth HP:0002714 |
| Epicanthus HP:0000286 |
| Feeding difficulties HP:0011968 |
| Gastroesophageal reflux HP:0002020 |
| Growth delay HP:0001510 |
| Low-set, posteriorly rotated ears HP:0000368 |
| Microretrognathia HP:0000308 |
| Muscle stiffness HP:0003552 |
| Neonatal respiratory distress HP:0002643 |
| Patent ductus arteriosus HP:0001643 |
| Plagiocephaly HP:0001357 |
| Ptosis HP:0000508 |
| Seizures HP:0001250 |
| Short stature HP:0004322 |
| Strabismus HP:0000486 |
| Ventricular septal defect HP:0001629 |
| Occasional |
| Brachydactyly HP:0001156 |
| Cleft palate HP:0000175 |
| Cryptorchidism HP:0000028 |
| Dystonia HP:0001332 |
| Hydronephrosis HP:0000126 |
| Intestinal malrotation HP:0002566 |
| Lacrimal duct stenosis HP:0007678 |
| Laryngomalacia HP:0001601 |
| Optic atrophy HP:0000648 |
| Preauricular pit HP:0004467 |
| Clinical Signs and Symptoms |
|---|
| HP:0000219 Thin (thick in the early age) upper lip vermilion |
| HP:0000341 Bitemporal narrowing |
| HP:0000341 Narrow forehead |
| HP:0000368 Low-set posteriorly rotated ears |
| HP:0000368 Low-set, posteriorly rotated ears |
| HP:0000391 Thickened helices |
| HP:0000414 Bulbous nose |
| HP:0000431 Wide nasal bridge |
| HP:0000486 Strabismus |
| HP:0000286 Epicanthus/HP:0000506 Telecanthus |
| HP:0001156 Brachydactyly |
| HP:0001263 Global developmental delay |
| HP:0001319 Neonatal hypotonia |
| HP:0001510 Growth delay |
| HP:0001999 Abnormal facial shape |
| HP:0002003 Large forehead |
| HP:0002020 Gastroesophageal reflux |
| HP:0002509 Limb hypertonia |
| HP:0002553 Highly arched eyebrows |
| HP:0002566 Intestinal malrotation/stenosis of sigma-rectum junction |
| HP:0002465 Poor speech/HP:0001344 Absent speech |
| HP:0004322 Short stature |
| HP:0008936 Muscular hypotonia of the trunk |
| HP:0010864 Intellectual disability, severe |
| HP:0011968 Feeding difficulties |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukvic, N.; Chetta, M.; Bagnulo, R.; Leotta, V.; Pantaleo, A.; Palumbo, O.; Palumbo, P.; Oro, M.; Rivieccio, M.; Laforgia, N.; et al. What Have We Learned from Patients Who Have Arboleda-Tham Syndrome Due to a De Novo KAT6A Pathogenic Variant with Impaired Histone Acetyltransferase Function? A Precise Clinical Description May Be Critical for Genetic Testing Approach and Final Diagnosis. Genes 2023, 14, 165. https://doi.org/10.3390/genes14010165
Bukvic N, Chetta M, Bagnulo R, Leotta V, Pantaleo A, Palumbo O, Palumbo P, Oro M, Rivieccio M, Laforgia N, et al. What Have We Learned from Patients Who Have Arboleda-Tham Syndrome Due to a De Novo KAT6A Pathogenic Variant with Impaired Histone Acetyltransferase Function? A Precise Clinical Description May Be Critical for Genetic Testing Approach and Final Diagnosis. Genes. 2023; 14(1):165. https://doi.org/10.3390/genes14010165
Chicago/Turabian StyleBukvic, Nenad, Massimiliano Chetta, Rosanna Bagnulo, Valentina Leotta, Antonino Pantaleo, Orazio Palumbo, Pietro Palumbo, Maria Oro, Maria Rivieccio, Nicola Laforgia, and et al. 2023. "What Have We Learned from Patients Who Have Arboleda-Tham Syndrome Due to a De Novo KAT6A Pathogenic Variant with Impaired Histone Acetyltransferase Function? A Precise Clinical Description May Be Critical for Genetic Testing Approach and Final Diagnosis" Genes 14, no. 1: 165. https://doi.org/10.3390/genes14010165
APA StyleBukvic, N., Chetta, M., Bagnulo, R., Leotta, V., Pantaleo, A., Palumbo, O., Palumbo, P., Oro, M., Rivieccio, M., Laforgia, N., De Rinaldis, M., Rosati, A., Kerkhof, J., Sadikovic, B., & Resta, N. (2023). What Have We Learned from Patients Who Have Arboleda-Tham Syndrome Due to a De Novo KAT6A Pathogenic Variant with Impaired Histone Acetyltransferase Function? A Precise Clinical Description May Be Critical for Genetic Testing Approach and Final Diagnosis. Genes, 14(1), 165. https://doi.org/10.3390/genes14010165