
Genetic Influences on Fetal Alcohol Spectrum Disorder
Abstract
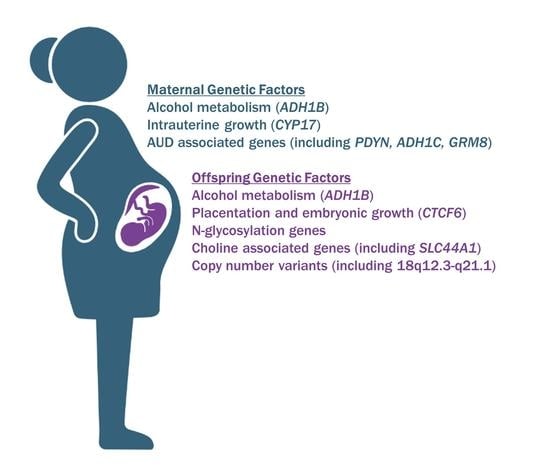
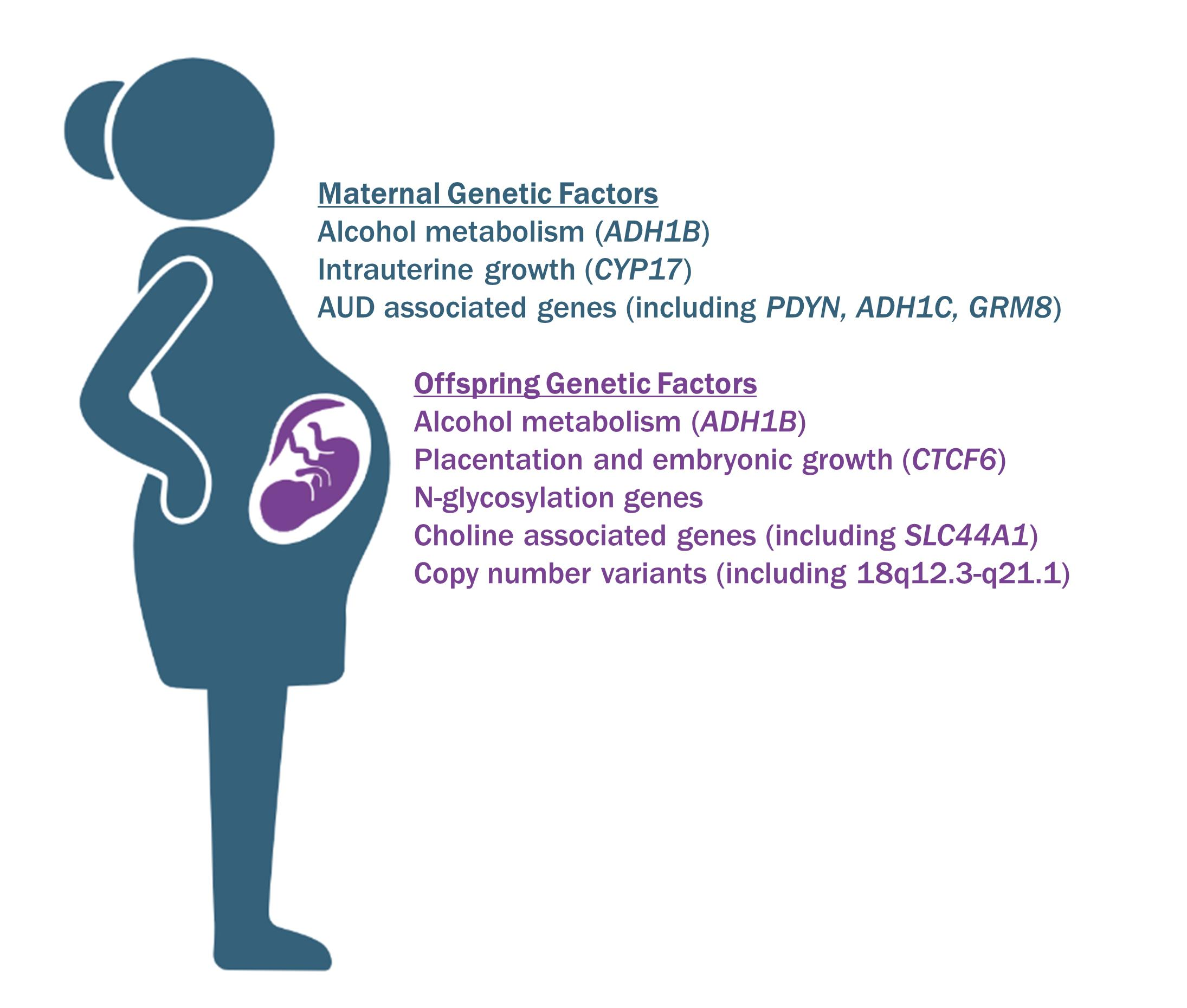
:
1. Introduction
2. Discordance and Concordance in Twin Studies
2.1. Alcohol-Metabolizing Enzymes
2.2. Other Genes and Pathways
2.3. Identification of Copy Number Variations by Genetic Testing
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bailey, B.A.; Sokol, R.J. Prenatal alcohol exposure and miscarriage, stillbirth, preterm delivery, and sudden infant death syndrome. Alcohol Res. Health 2011, 34, 86–91. [Google Scholar]
- Hoyme, H.E.; Kalberg, W.O.; Elliott, A.J.; Blankenship, J.; Buckley, D.; Marais, A.S.; Manning, M.A.; Robinson, L.K.; Adam, M.P.; Abdul-Rahman, O.; et al. Updated Clinical Guidelines for Diagnosing Fetal Alcohol Spectrum Disorders. Pediatrics 2016, 138, e20154256. [Google Scholar] [CrossRef] [Green Version]
- May, P.A.; Chambers, C.D.; Kalberg, W.O.; Zellner, J.; Feldman, H.; Buckley, D.; Kopald, D.; Hasken, J.M.; Xu, R.; Honerkamp-Smith, G.; et al. Prevalence of Fetal Alcohol Spectrum Disorders in 4 US Communities. JAMA 2018, 319, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Lange, S.; Probst, C.; Gmel, G.; Rehm, J.; Burd, L.; Popova, S. Global Prevalence of Fetal Alcohol Spectrum Disorder Among Children and Youth: A Systematic Review and Meta-analysis. JAMA Pediatr. 2017, 171, 948–956. [Google Scholar] [CrossRef]
- Bakhireva, L.N.; Garrison, L.; Shrestha, S.; Sharkis, J.; Miranda, R.; Rogers, K. Challenges of diagnosing fetal alcohol spectrum disorders in foster and adopted children. Alcohol 2018, 67, 37–43. [Google Scholar] [CrossRef]
- Morleo, M.; Woolfall, K.; Dedman, D.; Mukherjee, R.; Bellis, M.A.; Cook, P.A. Under-reporting of foetal alcohol spectrum disorders: An analysis of hospital episode statistics. BMC Pediatr. 2011, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Popova, S.; Lange, S.; Shield, K.; Mihic, A.; Chudley, A.E.; Mukherjee, R.A.S.; Bekmuradov, D.; Rehm, J. Comorbidity of fetal alcohol spectrum disorder: A systematic review and meta-analysis. Lancet 2016, 387, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Stoler, J.M.; Holmes, L.B. Under-recognition of prenatal alcohol effects in infants of known alcohol abusing women. J. Pediatr. 1999, 135, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.L. An update on incidence of FAS: FAS is not an equal opportunity birth defect. Neurotoxicol. Teratol. 1995, 17, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Wattendorf, D.J.; Muenke, M. Fetal alcohol spectrum disorders. Am. Fam. Physician 2005, 72, 279–282, 285. [Google Scholar]
- Green, R.F.; Stoler, J.M. Alcohol dehydrogenase 1B genotype and fetal alcohol syndrome: A HuGE minireview. Am. J. Obstet. Gynecol. 2007, 197, 12–25. [Google Scholar] [CrossRef] [Green Version]
- May, P.A.; Gossage, J.P. Maternal risk factors for fetal alcohol spectrum disorders: Not as simple as it might seem. Alcohol Res. Health 2011, 34, 15–26. [Google Scholar] [PubMed]
- Keen, C.L.; Uriu-Adams, J.Y.; Skalny, A.; Grabeklis, A.; Grabeklis, S.; Green, K.; Yevtushok, L.; Wertelecki, W.W.; Chambers, C.D. The plausibility of maternal nutritional status being a contributing factor to the risk for fetal alcohol spectrum disorders: The potential influence of zinc status as an example. Biofactors 2010, 36, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingol, N.; Schuster, C.; Fuchs, M.; Iosub, S.; Turner, G.; Stone, R.K.; Gromisch, D.S. The influence of socioeconomic factors on the occurrence of fetal alcohol syndrome. Adv. Alcohol Subst. Abus. 1987, 6, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, J.K.; Parnell, S.E. The Genetics of Fetal Alcohol Spectrum Disorders. Alcohol. Clin. Exp. Res. 2016, 40, 1154–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debelak, K.A.; Smith, S.M. Avian genetic background modulates the neural crest apoptosis induced by ethanol exposure. Alcohol. Clin. Exp. Res. 2000, 24, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Flink, S.; Florez-McClure, M.L.; Johnson, T.E.; Tabakoff, B.; Kechris, K.J. Gene expression changes in C57BL/6J and DBA/2J mice following prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2012, 36, 1519–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.L.; Singh, A.V.; Zhang, Y.; Nemeth, K.A.; Sulik, K.K.; Knudsen, T.B. Reprogramming of genetic networks during initiation of the Fetal Alcohol Syndrome. Dev. Dyn. 2007, 236, 613–631. [Google Scholar] [CrossRef]
- Mead, E.A.; Sarkar, D.K. Fetal alcohol spectrum disorders and their transmission through genetic and epigenetic mechanisms. Front. Genet. 2014, 5, 154. [Google Scholar] [CrossRef] [Green Version]
- Kaminen-Ahola, N. Fetal alcohol spectrum disorders: Genetic and epigenetic mechanisms. Prenat. Diagn. 2020, 40, 1185–1192. [Google Scholar] [CrossRef]
- Christoffel, K.K.; Salafsky, I. Fetal alcohol syndrome in dizygotic twins. J. Pediatr. 1975, 87, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Chasnoff, I.J. Fetal alcohol syndrome in twin pregnancy. Acta Genet. Med. Gemellol. 1985, 34, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Riese, M.L. Maternal alcohol and pentazocine abuse: Neonatal behavior and morphology in an opposite-sex twin pair. Acta Genet. Med. Gemellol. 1989, 38, 49–56. [Google Scholar] [CrossRef]
- Riikonen, R.S. Difference in susceptibility to teratogenic effects of alcohol in discordant twins exposed to alcohol during the second half of gestation. Pediatr. Neurol. 1994, 11, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Streissguth, A.P.; Dehaene, P. Fetal alcohol syndrome in twins of alcoholic mothers: Concordance of diagnosis and IQ. Am. J. Med. Genet. 1993, 47, 857–861. [Google Scholar] [CrossRef]
- Astley Hemingway, S.J.; Bledsoe, J.M.; Brooks, A.; Davies, J.K.; Jirikowic, T.; Olson, E.M.; Thorne, J.C. Twin study confirms virtually identical prenatal alcohol exposures can lead to markedly different fetal alcohol spectrum disorder outcomes-fetal genetics influences fetal vulnerability. Adv. Pediatr. Res. 2018, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.; Oullette, E.M.; Warner, L.; Leichtman, S.R. Congenital malformations in offspring of a chronic alcoholic mother. Pediatrics 1974, 53, 490–494. [Google Scholar] [CrossRef]
- Gelernter, J.; Kranzler, H.R. Genetics of alcohol dependence. Hum. Genet. 2009, 126, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Polimanti, R.; Gelernter, J. ADH1B: From alcoholism, natural selection, and cancer to the human phenome. Am. J. Med. Genet B Neuropsychiatr. Genet. 2018, 177, 113–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 2007, 30, 5–13. [Google Scholar]
- Hurley, T.D.; Edenberg, H.J. Genes encoding enzymes involved in ethanol metabolism. Alcohol Res. 2012, 34, 339–344. [Google Scholar]
- Eng, M.Y.; Luczak, S.E.; Wall, T.L. ALDH2, ADH1B, and ADH1C genotypes in Asians: A literature review. Alcohol Res. Health 2007, 30, 22–27. [Google Scholar]
- McCarthy, D.M.; Pedersen, S.L.; Lobos, E.A.; Todd, R.D.; Wall, T.L. ADH1B*3 and response to alcohol in African-Americans. Alcohol. Clin. Exp. Res. 2010, 34, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Crabb, D.W.; Edenberg, H.J.; Bosron, W.F.; Li, T.K. Genotypes for aldehyde dehydrogenase deficiency and alcohol sensitivity. The inactive ALDH2(2) allele is dominant. J. Clin. Investig. 1989, 83, 314–316. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.J.; Enoch, M.A.; Goldman, D.; Li, T.K.; Yokoyama, A. The alcohol flushing response: An unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009, 6, e50. [Google Scholar] [CrossRef]
- Khaole, N.C.; Ramchandani, V.A.; Viljoen, D.L.; Li, T.K. A pilot study of alcohol exposure and pharmacokinetics in women with or without children with fetal alcohol syndrome. Alcohol Alcohol. 2004, 39, 503–508. [Google Scholar] [CrossRef]
- Smith, M.; Hopkinson, D.A.; Harris, H. Developmental changes and polymorphism in human alcohol dehydrogenase. Ann. Hum. Genet. 1971, 34, 251–271. [Google Scholar] [CrossRef] [Green Version]
- Pikkarainen, P.H.; Raiha, N.C. Development of alcohol dehydrogenase activity in the human liver. Pediatr. Res. 1967, 1, 165–168. [Google Scholar] [CrossRef] [Green Version]
- McCarver, D.G.; Thomasson, H.R.; Martier, S.S.; Sokol, R.J.; Li, T. Alcohol dehydrogenase-2*3 allele protects against alcohol-related birth defects among African Americans. J. Pharmacol. Exp. Ther. 1997, 283, 1095–1101. [Google Scholar]
- Das, U.G.; Cronk, C.E.; Martier, S.S.; Simpson, P.M.; McCarver, D.G. Alcohol dehydrogenase 2*3 affects alterations in offspring facial morphology associated with maternal ethanol intake in pregnancy. Alcohol. Clin. Exp. Res. 2004, 28, 1598–1606. [Google Scholar] [CrossRef]
- Jacobson, S.W.; Carr, L.G.; Croxford, J.; Sokol, R.J.; Li, T.K.; Jacobson, J.L. Protective effects of the alcohol dehydrogenase-ADH1B allele in children exposed to alcohol during pregnancy. J. Pediatr. 2006, 148, 30–37. [Google Scholar] [CrossRef]
- Dodge, N.C.; Jacobson, J.L.; Jacobson, S.W. Protective effects of the alcohol dehydrogenase-ADH1B*3 allele on attention and behavior problems in adolescents exposed to alcohol during pregnancy. Neurotoxicol. Teratol. 2014, 41, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Stoler, J.M.; Ryan, L.M.; Holmes, L.B. Alcohol dehydrogenase 2 genotypes, maternal alcohol use, and infant outcome. J. Pediatr. 2002, 141, 780–785. [Google Scholar] [CrossRef]
- Viljoen, D.L.; Carr, L.G.; Foroud, T.M.; Brooke, L.; Ramsay, M.; Li, T.K. Alcohol dehydrogenase-2*2 allele is associated with decreased prevalence of fetal alcohol syndrome in the mixed-ancestry population of the Western Cape Province, South Africa. Alcohol. Clin. Exp. Res. 2001, 25, 1719–1722. [Google Scholar] [CrossRef]
- Zuccolo, L.; Fitz-Simon, N.; Gray, R.; Ring, S.M.; Sayal, K.; Smith, G.D.; Lewis, S.J. A non-synonymous variant in ADH1B is strongly associated with prenatal alcohol use in a European sample of pregnant women. Hum. Mol. Genet. 2009, 18, 4457–4466. [Google Scholar] [CrossRef] [Green Version]
- Lewis, S.J.; Zuccolo, L.; Davey Smith, G.; Macleod, J.; Rodriguez, S.; Draper, E.S.; Barrow, M.; Alati, R.; Sayal, K.; Ring, S.; et al. Fetal alcohol exposure and IQ at age 8: Evidence from a population-based birth-cohort study. PLoS ONE 2012, 7, e49407. [Google Scholar] [CrossRef]
- Murray, J.; Burgess, S.; Zuccolo, L.; Hickman, M.; Gray, R.; Lewis, S.J. Moderate alcohol drinking in pregnancy increases risk for children’s persistent conduct problems: Causal effects in a Mendelian randomisation study. J. Child Psychol. Psychiatry 2016, 57, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Delpisheh, A.; Topping, J.; Reyad, M.; Tang, A.; Brabin, B.J. Prenatal alcohol exposure, CYP17 gene polymorphisms and fetal growth restriction. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 138, 49–53. [Google Scholar] [CrossRef]
- Marjonen, H.; Kahila, H.; Kaminen-Ahola, N. rs10732516 polymorphism at the IGF2/H19 locus associates with a genotype-specific trend in placental DNA methylation and head circumference of prenatally alcohol-exposed newborns. Hum. Reprod. Open 2017, 2017, hox014. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Virdee, M.S.; Eckerle, J.K.; Sandness, K.E.; Georgieff, M.K.; Boys, C.J.; Zeisel, S.H.; Wozniak, J.R. Polymorphisms in SLC44A1 are associated with cognitive improvement in children diagnosed with fetal alcohol spectrum disorder: An exploratory study of oral choline supplementation. Am. J. Clin. Nutr. 2021, 114, 617–627. [Google Scholar] [CrossRef]
- de la Morena-Barrio, M.E.; Ballesta-Martinez, M.J.; Lopez-Galvez, R.; Anton, A.I.; Lopez-Gonzalez, V.; Martinez-Ribot, L.; Padilla, J.; Minano, A.; Garcia-Algar, O.; Del Campo, M.; et al. Genetic predisposition to fetal alcohol syndrome: Association with congenital disorders of N-glycosylation. Pediatr. Res. 2018, 83, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Wehby, G.L.; Prater, K.N.; Ryckman, K.K.; Kummet, C.; Murray, J.C. Candidate gene study for smoking, alcohol use, and body weight in a sample of pregnant women. J. Matern. Fetal Neonatal Med. 2015, 28, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Ukita, K.; Fukui, Y.; Shiota, K. Effects of prenatal alcohol exposure in mice: Influence of an ADH inhibitor and a chronic inhalation study. Reprod. Toxicol. 1993, 7, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Webster, W.S.; Walsh, D.A.; McEwen, S.E.; Lipson, A.H. Some teratogenic properties of ethanol and acetaldehyde in C57BL/6J mice: Implications for the study of the fetal alcohol syndrome. Teratology 1983, 27, 231–243. [Google Scholar] [CrossRef]
- Reimers, M.J.; Flockton, A.R.; Tanguay, R.L. Ethanol- and acetaldehyde-mediated developmental toxicity in zebrafish. Neurotoxicol. Teratol. 2004, 26, 769–781. [Google Scholar] [CrossRef]
- Heller, M.; Burd, L. Review of ethanol dispersion, distribution, and elimination from the fetal compartment. Birth Defects Res. A Clin. Mol. Teratol. 2014, 100, 277–283. [Google Scholar] [CrossRef]
- Burd, L.; Blair, J.; Dropps, K. Prenatal alcohol exposure, blood alcohol concentrations and alcohol elimination rates for the mother, fetus and newborn. J. Perinatol. 2012, 32, 652–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benonisdottir, S.; Oddsson, A.; Helgason, A.; Kristjansson, R.P.; Sveinbjornsson, G.; Oskarsdottir, A.; Thorleifsson, G.; Davidsson, O.B.; Arnadottir, G.A.; Sulem, G.; et al. Epigenetic and genetic components of height regulation. Nat. Commun. 2016, 7, 13490. [Google Scholar] [CrossRef] [Green Version]
- Savage, J.E.; Salvatore, J.E.; Aliev, F.; Edwards, A.C.; Hickman, M.; Kendler, K.S.; Macleod, J.; Latvala, A.; Loukola, A.; Kaprio, J.; et al. Polygenic Risk Score Prediction of Alcohol Dependence Symptoms Across Population-Based and Clinically Ascertained Samples. Alcohol. Clin. Exp. Res. 2018, 42, 520–530. [Google Scholar] [CrossRef]
- Douzgou, S.; Breen, C.; Crow, Y.J.; Chandler, K.; Metcalfe, K.; Jones, E.; Kerr, B.; Clayton-Smith, J. Diagnosing fetal alcohol syndrome: New insights from newer genetic technologies. Arch. Dis. Child. 2012, 97, 812–817. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Abdelmalik, N.; van Haelst, M.; Mancini, G.; Schrander-Stumpel, C.; Marcus-Soekarman, D.; Hennekam, R.; Cobben, J.M. Diagnostic outcomes of 27 children referred by pediatricians to a genetics clinic in the Netherlands with suspicion of fetal alcohol spectrum disorders. Am. J. Med. Genet. A 2013, 161A, 254–260. [Google Scholar] [CrossRef]
- Jamuar, S.S.; Picker, J.D.; Stoler, J.M. Utility of Genetic Testing in Fetal Alcohol Spectrum Disorder. J. Pediatr. 2018, 196, 270–274.e271. [Google Scholar] [CrossRef]
- Zarrei, M.; Hicks, G.G.; Reynolds, J.N.; Thiruvahindrapuram, B.; Engchuan, W.; Pind, M.; Lamoureux, S.; Wei, J.; Wang, Z.; Marshall, C.R.; et al. Copy number variation in fetal alcohol spectrum disorder. Biochem. Cell. Biol. 2018, 96, 161–166. [Google Scholar] [CrossRef]
- Lam, Z.; Johnson, K.; Jewell, R. Genetic testing in patients with possible foetal alcohol spectrum disorder. Arch. Dis. Child. 2021, 106, 653–655. [Google Scholar] [CrossRef]
- Kahila, H.; Marjonen, H.; Auvinen, P.; Avela, K.; Riikonen, R.; Kaminen-Ahola, N. 18q12.3-q21.1 microdeletion detected in the prenatally alcohol-exposed dizygotic twin with discordant fetal alcohol syndrome phenotype. Mol. Genet. Genomic. Med. 2020, 8, e1192. [Google Scholar] [CrossRef]
- Bailey, J.A.; Kidd, J.M.; Eichler, E.E. Human copy number polymorphic genes. Cytogenet. Genome Res. 2008, 123, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Bingol, N.; Fuchs, M.; Iosub, S.; Kumar, S.; Stone, R.K.; Gromisch, D.S. Fetal alcohol syndrome associated with trisomy 21. Alcohol. Clin. Exp. Res. 1987, 11, 42–44. [Google Scholar] [CrossRef]
- Romke, C.; Heyne, K.; Stewens, J.; Schwinger, E. Erroneous diagnosis of fetal alcohol syndrome in a patient with ring chromosome 6. Eur. J. Pediatr. 1987, 146, 443. [Google Scholar] [CrossRef]
- Muller, J.; Cobet, G.; Laske, G.; Degen, B.; Grauel, C.; Lehmann, K. [Partial monosomy 21 or fetal alcohol embryopathy in a retarded boy?]. Padiatr. Grenzgeb. 1993, 31, 313–319. [Google Scholar]
- Weyrauch, D.; Schwartz, M.; Hart, B.; Klug, M.G.; Burd, L. Comorbid Mental Disorders in Fetal Alcohol Spectrum Disorders: A Systematic Review. J. Dev. Behav. Pediatr. 2017, 38, 283–291. [Google Scholar] [CrossRef]
- Xu, G.; Strathearn, L.; Liu, B.; Yang, B.; Bao, W. Twenty-Year Trends in Diagnosed Attention-Deficit/Hyperactivity Disorder Among US Children and Adolescents, 1997–2016. JAMA Netw. Open 2018, 1, e181471. [Google Scholar] [CrossRef] [Green Version]
- Biederman, J.; Faraone, S.V. Attention-deficit hyperactivity disorder. Lancet 2005, 366, 237–248. [Google Scholar] [CrossRef]
- Pedersen, S.L.; Walther, C.A.; Harty, S.C.; Gnagy, E.M.; Pelham, W.E.; Molina, B.S. The indirect effects of childhood attention deficit hyperactivity disorder on alcohol problems in adulthood through unique facets of impulsivity. Addiction 2016, 111, 1582–1589. [Google Scholar] [CrossRef]

{kind=link}
| Polymorphism | N | Findings | Source |
|---|---|---|---|
| Alcohol dehydrogenase (ADH) | |||
| AHD1B Cys370 (ADH1B*3) | 247 African American mother–offspring pairs | Improved development in infants of drinking mothers carrying Cys370 Protective effect of Cys370 against PAE-induced facial dysmorphology | McCarver et al., 1997 [39] Das et al., 2004 [40] |
| 263 African American mother–offspring pairs | Protective effects of Cys370 on birth size as well as behavioral and cognitive outcomes at infancy and 7.5 years Cys370 carriers were less likely to have hyperactivity and inattention at 14 years | Jacobson et al., 2006 [41] Dodge et al., 2014 [42] | |
| 404 mixed-race women and 139 infants | Cys370 carrier mothers had increased risk for growth restriction and/or facial dysmorphia; Cys370 was more frequent in affected infants | Stoler et al., 2002 [43] | |
| ADH1B His48 (ADH1B*2) | 56 mixed-ancestry mother–offspring pairs | Lower frequency of His48 in FAS children and their mothers | Viljoen, et al., 2001 [44] |
| ADH1B rs1229984 | 7410 white European women | Associated with alcohol consumption during pregnancy | Zuccolo et al., 2009 [45] |
| ADH1A rs2866151 ADH1A rs975833 ADH1B rs4147536 ADH7 rs284779 | 6196 children of white European women | These 4 ADH loci of 10 tested were associated with lower IQ in children of mothers who drank moderately during pregnancy | Lewis et al., 2012 [46] |
| 3500 children of white European women | The same 4 ADH loci predicted increased risk of early-onset persistent conduct problems among children of mothers who drank moderately during pregnancy | Murray et al., 2016 [47] | |
| Other Genes | |||
| CYP17 A1 allele | 180 controls and 90 cases, Caucasian women | Associated with intrauterine growth restriction in infants of mothers who consumed alcohol during pregnancy | Delpisheh et al., 2008 [48] |
| CTCF6 rs10732516 | 100 Caucasian controls and 39 predominately Caucasian alcohol-exposed | Associated with decreased newborn head circumference and decreased methylation at the H19 imprinting coding region (ICR) of IGF2 in alcohol-exposed placentas | Marjonen et al., 2017 [49] |
| SLC44A1 | 52 children diagnosed with FASD | 14 SNPs in SLC44A1 were associated with increased cognitive performance after choline supplementation | Smith et al., 2021 [50] |
| N-glycosylation genes | 20 controls and 25 FAS patients of different European ancestries | Rare variants in genes associated with N-glycosylation were more frequent in FAS patients | de la Morena-Barrio et al., 2018 [51] |
| PYDN, ADH1C, and GRM8 | 1937 mothers of primarily Caucasian background | SNPs in 39 genes (including PYDN, ADH1C, and GRM8) were associated with drinking during different stages of pregnancy | Wehby et al., 2015 [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sambo, D.; Goldman, D. Genetic Influences on Fetal Alcohol Spectrum Disorder. Genes 2023, 14, 195. https://doi.org/10.3390/genes14010195
Sambo D, Goldman D. Genetic Influences on Fetal Alcohol Spectrum Disorder. Genes. 2023; 14(1):195. https://doi.org/10.3390/genes14010195
Chicago/Turabian StyleSambo, Danielle, and David Goldman. 2023. "Genetic Influences on Fetal Alcohol Spectrum Disorder" Genes 14, no. 1: 195. https://doi.org/10.3390/genes14010195
APA StyleSambo, D., & Goldman, D. (2023). Genetic Influences on Fetal Alcohol Spectrum Disorder. Genes, 14(1), 195. https://doi.org/10.3390/genes14010195