
Fitting Genomic Prediction Models with Different Marker Effects among Prefectures to Carcass Traits in Japanese Black Cattle

Abstract
:1. Introduction
2. Materials and Methods
2.1. Theory
2.2. Data Analysis
3. Results
3.1. Variance Component Estimation
3.2. SNP Effects and Genomic Breeding Values
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goddard, M.E.; Hayes, B.J. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 2009, 10, 381–391. [Google Scholar] [CrossRef]
- Brøndum, R.F.; Rius-Vilarrasa, E.; Strandén, I.; Su, G.; Guldbrandtsen, B.; Fikse, W.F.; Lund, M.S. Reliabilities of genomic prediction using combined reference data of the Nordic Red dairy cattle populations. J. Dairy Sci. 2011, 94, 4700–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, M.S.; de Roos, A.P.W.; de Vries, A.G.; Druet, T.; Ducrocq, V.; Fritz, S.; Guillaume, F.; Guldbrandtsen, B.; Liu, Z.; Reents, R.; et al. A common reference population from four European Holstein populations increases reliability of genomic predictions. Genet. Sel. Evol. 2011, 43, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heringstad, B.; Su, G.; Solberg, T.R.; Guldbrandtsen, B.; Svendsen, M.; Lund, M.S. Genomic predictions based on a joint ref-erence population for Scandinavian red breeds. In Proceedings of the 62nd Annual Meeting of the European Federation of Animal Science, Stavanger, Norway, 29 August–2 September 2011. [Google Scholar]
- de Roos, A.P.W.; Hayes, B.J.; Goddard, M.E. Reliability of genomic predictions across multiple populations. Genetics 2009, 183, 1545–1553. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.C.; Verbyla, K.; Goddard, M.E. Accuracy of genomic breeding values in multi-breed dairy cattle populations. Genet. Sel. Evol. 2009, 41, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, M.S.; Su, G.; Janss, L.; Guldbrandtsen, B.; Brøndum, R.F. Genomic evaluation of cattle in a multi-breed context. Livest. Sci. 2014, 166, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Karoui, S.; Carabaño, M.J.; Díaz, C.; Legarra, A. Joint genomic evaluation of French dairy cattle breeds using multiple-trait models. Genet. Sel. Evol. 2012, 44, 39. [Google Scholar] [CrossRef] [Green Version]
- Thomasen, J.R.; Sørensen, A.C.; Su, G.; Madsen, P.; Lund, M.S.; Guldbrandtsen, B. The admixed population structure in Danish Jersey dairy cattle challenges accurate genomic predictions. J. Anim. Sci. 2013, 91, 3105–3112. [Google Scholar] [CrossRef] [Green Version]
- Khansefid, M.; Pryce, J.E.; Bolormaa, S.; Miller, S.P.; Wang, Z.; Li, C.; Goddard, M.E. Estimation of genomic breeding values for residual feed intake in a multibreed cattle population. J. Anim. Sci. 2014, 92, 3270–3283. [Google Scholar] [CrossRef] [Green Version]
- Iheshiulor, O.O.; Woolliams, J.A.; Yu, X.; Wellmann, R.; Meuwissen, T.H. Within- and across-breed genomic prediction using whole-genome sequence and single nucleotide polymorphism panels. Genet. Sel. Evol. 2016, 48, 15. [Google Scholar] [CrossRef]
- MacLeod, I.M.; Bowman, P.J.; Vander Jagt, C.J.; Haile-Mariam, M.; Kemper, K.E.; Chamberlain, A.J.; Schrooten, C.; Hayes, B.J.; Goddard, M.E. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 144. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, T.; van den Berg, I.; Goddard, M. On the use of whole-genome sequence data for across-breed genomic prediction and fine-scale mapping of QTL. Genet. Sel. Evol. 2021, 53, 19. [Google Scholar] [CrossRef]
- Varona, L.; Moreno, C.; Ibañez-Escriche, N.; Altarriba, J. Whole genome evaluation for related populations. In Proceedings of the 9th World Congress on Genetics Applied to Livestock Production, Leipzig, Germany, 1–6 August 2010. [Google Scholar]
- Makgahlela, M.L.; Strandén, I.; Nielsen, U.S.; Sillanpää, M.J.; Mäntysaari, E.A. The estimation of genomic relationships using breedwise allele frequencies among animals in multibreed populations. J. Dairy Sci. 2013, 96, 5364–5375. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Matsuda, H.; Taniguchi, Y.; Watanabe, T.; Tabuchi, I.; Iwaisaki, H. An attempt of genomic prediction for carcass traits in Japanese Black cattle using a statistical model considering QTL × subpopulation interaction effect. In Proceedings of the 16th Meeting of the Japanese Society of Animal Breeding and Genetics, Kobe, Japan, 6–8 November 2015. (In Japanese). [Google Scholar]
- Namikawa, K. Wagyu: Japanese Beef Cattle—Historical Breeding Processes of Japanese Beef Cattle and Preservation of Genetic Resources as Economic Farm Animal; Wagyu Registry Association: Kyoto, Japan, 1992. [Google Scholar]
- Gotoh, T.; Takahashi, H.; Nishimura, T.; Kuchida, K.; Mannen, H. Meat produced by Japanese Black cattle and Wagyu. Anim. Front. 2014, 4, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Motoyama, M.; Sasaki, K.; Watanabe, A. Wagyu and the factors contributing to its beef quality: A Japanese industry overview. Meat Sci. 2016, 120, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Ashida, I.; Iwaisaki, H. A numerical technique for REML estimation of variance components using average information algorithm and its computing property. Anim. Sci. Technol. 1998, 69, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Ashida, I.; Iwaisaki, H. An expression for average information matrix for a mixed linear multi-component of variance model and REML iteration equations. Anim. Sci. J. 1999, 70, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Wagyu Registry Association. Breeding and Improvement of Wagyu; Wagyu Registry Association: Kyoto, Japan, 2007. (In Japanese) [Google Scholar]
- Nishimaki, T.; Ibi, T.; Tanabe, Y.; Miyazaki, Y.; Kobayashi, N.; Matsuhashi, T.; Akiyama, T.; Yoshida, E.; Imai, K.; Matsui, M.; et al. The assessment of genetic diversity within and among the eight subpopulations of Japanese Black cattle using 52 microsatellite markers. Anim. Sci. J. 2013, 84, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Suezawa, R.; Nikadori, H.; Sasaki, S. Genetic diversity and genomic inbreeding in Japanese Black cows in the islands of Okinawa Prefecture evaluated using single-nucleotide polymorphism array. Anim. Sci. J. 2021, 92, e13525. [Google Scholar] [CrossRef]
- Komiya, R.; Ogawa, S.; Aonuma, T.; Satoh, M. Performance of using opposing homozygotes for paternity testing in Japanese Black cattle. J. Anim. Breed. Genet. 2022, 139, 113–124. [Google Scholar] [CrossRef]
- Zoda, A.; Ogawa, S.; Matsuda, H.; Taniguchi, Y.; Watanabe, T.; Sugimoto, Y.; Iwaisaki, H. Inferring genetic characteristics of Japanese Black cattle populations using genome-wide single nucleotide polymorphism markers. J. Anim. Genet. 2022, 50, 3–9. [Google Scholar] [CrossRef]
- Kawaguchi, F.; Nakamura, M.; Kobayashi, E.; Yonezawa, T.; Sasazaki, S. Comprehensive assessment of genetic diversity, structure, and relationship in four Japanese cattle breeds by Illumina 50 K SNP array analysis. Anim. Sci. J. 2022, 93, e13770. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Matsuda, H.; Arakawa, A.; Yamada, T.; Iwaisaki, H.; Nishimura, S.; Sugimoto, Y. Estimation of variance components for carcass traits in Japanese Black cattle using 50K SNP genotype data. Anim. Sci. J. 2014, 85, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Matsuda, H.; Taniguchi, Y.; Watanabe, T.; Nishimura, S.; Sugimoto, Y.; Iwaisaki, H. Effects of single nucleotide polymorphism marker density on degree of genetic variance explained and genomic evaluation for carcass traits in Japanese Black beef cattle. BMC Genet. 2014, 15, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onogi, A.; Ogino, A.; Komatsu, T.; Shoji, N.; Simizu, K.; Kurogi, K.; Yasumori, T.; Togashi, K.; Iwata, H. Genomic prediction in Japanese Black cattle: Application of a single-step approach to beef cattle. J. Anim. Sci. 2014, 92, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T. Genomic breeding value evaluation for economically important traits of Japanese Black cattle. J. Anim. Genet. 2016, 44, 3–10. (In Japanese) [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Inoue, K.; Oyama, H.; Uchiyama, K.; Yoshinari, K.; Sasago, N.; Kojima, T.; Kashima, M.; Suzuki, H.; Kamata, T.; et al. Exploring the size of reference population for expected accuracy of genomic prediction using simulated and real data in Japanese Black cattle. BMC Genom. 2021, 22, 799. [Google Scholar] [CrossRef]
- Ogawa, S.; Matsuda, H.; Taniguchi, Y.; Watanabe, T.; Kitamura, Y.; Tabuchi, I.; Sugimoto, Y.; Iwaisaki, H. Genomic prediction for carcass traits in Japanese Black cattle using single nucleotide polymorphism markers of different densities. Anim. Prod. Sci. 2016, 57, 1631–1636. [Google Scholar] [CrossRef]
- Hirano, T.; Nishimura, S.; Watanabe, T.; Takasuga, A.; Hanzawa, K.; Sugimoto, Y. SNP discovery and evaluation with whole genome re-sequencing using pooled DNA in Japanese Black cattle. Nihon Chikusan Gakkaiho 2013, 84, 319–325. (In Japanese) [Google Scholar] [CrossRef]
- Sasaki, S.; Watanabe, T.; Nishimura, S.; Sugimoto, Y. Genome-wide identification of copy number variation using high-density single-nucleotide polymorphism array in Japanese Black cattle. BMC Genet. 2016, 17, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, S.; Watanabe, T.; Ibi, T.; Hasegawa, K.; Sakamoto, Y.; Moriwaki, S.; Kurogi, K.; Ogino, A.; Yasumori, T.; Wakaguri, H.; et al. Identification of deleterious recessive haplotypes and candidate deleterious recessive mutations in Japanese Black cattle. Sci. Rep. 2021, 11, 6687. [Google Scholar] [CrossRef]
- Arishima, T.; Wakaguri, H.; Nakashima, R.; Sakakihara, S.; Kawashima, K.; Sugimoto, Y.; Suzuki, Y.; Sasaki, S. Comprehensive analysis of 124 transcriptomes from 31 tissues in developing, juvenile, and adult Japanese Black Cattle. DNA Res. 2022, 29, dsac032. [Google Scholar] [CrossRef]
- Zoda, A.; Ogawa, S.; Matsuda, H.; Taniguchi, Y.; Watanabe, T.; Sugimoto, Y.; Iwaisaki, H. Genomic prediction for carcass traits in Japanese Black cattle considering mixed structure of subpopulations. J. Anim. Genet. 2022, 50, 31–38. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- de los Campos, G.; Sorensen, D. On the genomic analysis of data from structured populations. J. Anim. Breed. Genet. 2014, 131, 163–164. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Campos, G.D.L.; VandeHaar, M.; Spurlock, D.; Armentano, L.; Coffey, M.; de Haas, Y.; Veerkamp, R.; Staples, C.; Connor, E.; et al. Use of genotype × environment interaction model to accommodate genetic heterogeneity for residual feed intake, dry matter intake, net energy in milk, and metabolic body weight in dairy cattle. J. Dairy Sci. 2017, 100, 2007–2016. [Google Scholar] [CrossRef] [Green Version]
- Legarra, A.; Baloche, G.; Barillet, F.; Astruc, J.; Soulas, C.; Aguerre, X.; Arrese, F.; Mintegi, L.; Lasarte, M.; Maeztu, F.; et al. Within- and across-breed genomic predictions and genomic relationships for Western Pyrenees dairy sheep breeds Latxa, Manech, and Basco-Béarnaise. J. Dairy Sci. 2014, 97, 3200–3212. [Google Scholar] [CrossRef] [Green Version]
- Japan Meat Grading Association. New Beef Carcass Grading Standards; Japan Meat Grading Association: Tokyo, Japan, 1988. [Google Scholar]
- Browning, B.L.; Browning, S.R. Haplotypic analysis of Wellcome Trust Case Control Consortium data. Hum. Genet. 2008, 123, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Pérez, P.; de los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Spiegelhalter, D.J.; Best, N.G.; Carlin, B.P.; van der Linde, A. Bayesian measures of model complexity and fit. J. R. Stat. Soc. Ser. B Stat. Methodol. 2002, 64, 583–639. [Google Scholar] [CrossRef] [Green Version]
- McClure, M.C.; Ramey, H.R.; Rolf, M.M.; McKay, S.D.; Decker, J.E.; Chapple, R.H.; Kim, J.W.; Taxis, T.M.; Weaber, R.L.; Schnabel, R.D.; et al. Genome-wide association analysis for quantitative trait loci influencing Warner-Bratzler shear force in five taurine cattle breeds. Anim. Genet. 2012, 43, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet. Res. 2012, 94, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Gualdrón Duarte, J.L.; Cantet, R.J.; Bates, R.O.; Ernst, C.W.; Raney, N.E.; Steibel, J.P. Rapid screening for phenotype-genotype associations by linear transformations of genomic evaluations. BMC Bioinform. 2014, 15, 246. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Matsuda, H.; Taniguchi, Y.; Watanabe, T.; Sugimoto, Y.; Iwaisaki, H. Estimation of the autosomal contribution to total additive genetic variability of carcass traits in Japanese Black cattle. Anim. Sci. J. 2022, 93, e13710. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Saito, H.; Satoh, M. Genetic relationship of female reproductive traits with calf weight and carcass traits in Japanese Black cattle population in Miyagi prefecture. Nihon Chikusan Gakkaiho 2022, 93, 97–104. (In Japanese) [Google Scholar] [CrossRef]
- Robertson, A. The sampling variance of the genetic correlation coefficient. Biometrics 1959, 15, 469–485. [Google Scholar] [CrossRef]
- Onogi, A.; Watanabe, T.; Ogino, A.; Kurogi, K.; Togashi, K. Genomic prediction with non-additive effects in beef cattle: Stability of variance component and genetic effect estimates against population size. BMC Genom. 2021, 22, 512. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Inoue, Y.; Oe, T.; Nishimura, M. Genomic imprinting variances of beef carcass traits and physiochemical characteristics in Japanese Black cattle. Anim. Sci. J. 2021, 92, e13504. [Google Scholar] [CrossRef]
- Karaman, E.; Su, G.; Croue, I.; Lund, M.S. Genomic prediction using a reference population of multiple pure breeds and admixed individuals. Genet. Sel. Evol. 2021, 53, 46. [Google Scholar] [CrossRef]
- Kudinov, A.A.; Mäntysaari, E.A.; Pitkänen, T.J.; Saksa, E.I.; Aamand, G.P.; Uimari, P.; Strandén, I. Single-step genomic evaluation of Russian dairy cattle using internal and external information. J. Anim. Breed. Genet. 2022, 139, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Lehermeier, C.; Schön, C.C.; de Los Campos, G. Assessment of genetic heterogeneity in structured plant populations using multivariate whole-genome regression models. Genetics 2015, 201, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Veturi, Y.; de Los Campos, G.; Yi, N.; Huang, W.; Vazquez, A.I.; Kühnel, B. Modeling heterogeneity in the genetic architecture of ethnically diverse groups using random effect interaction models. Genetics 2019, 211, 1395–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesinos-López, A.; Runcie, D.E.; Ibba, M.I.; Pérez-Rodríguez, P.; Montesinos-López, O.A.; Crespo, L.A.; Bentley, A.R.; Crossa, J. Multi-trait genomic-enabled prediction enhances accuracy in multi-year wheat breeding trials. G3 2021, 11, jkab270. [Google Scholar] [CrossRef] [PubMed]
- Steyn, Y.; Lourenco, D.A.L.; Misztal, I. Genomic predictions in purebreds with a multibreed genomic relationship matrix. J. Anim. Sci. 2019, 97, 4418–4427. [Google Scholar] [CrossRef]
- Nakaoka, H.; Narita, A.; Ibi, T.; Sasae, Y.; Miyake, T.; Yamada, T.; Sasaki, Y. Effectiveness of adjusting for heterogeneity of variance in genetic evaluation of Japanese Black cattle. J. Anim. Sci. 2007, 85, 2429–2436. [Google Scholar] [CrossRef]
- Nakaoka, H.; Gaillard, C.; Ibi, T.; Sasae, Y.; Sasaki, Y. Adjusting for heterogeneity of variance for carcass traits affects single and multiple trait selections in genetic evaluation of Japanese Black cattle. Anim. Sci. J. 2008, 79, 645–654. [Google Scholar] [CrossRef]
- Gebreyesus, G.; Lund, M.S.; Buitenhuis, B.; Bovenhuis, H.; Poulsen, N.A.; Janss, L.G. Modeling heterogeneous (co)variances from adjacent-SNP groups improves genomic prediction for milk protein composition traits. Genet. Sel. Evol. 2017, 49, 89. [Google Scholar] [CrossRef] [Green Version]
- Karaman, E.; Lund, M.S.; Anche, M.T.; Janss, L.; Su, G. Genomic prediction using multi-trait weighted GBLUP accounting for heterogeneous variances and covariances across the genome. G3 2018, 8, 3549–3558. [Google Scholar] [CrossRef] [Green Version]
- Karaman, E.; Lund, M.S.; Su, G. Multi-trait single-step genomic prediction accounting for heterogeneous (co)variances over the genome. Heredity 2020, 124, 274–287. [Google Scholar] [CrossRef]
- Nomura, T.; Sasaki, Y. Studies on genetic differentiation of Japanese Black cattle by means of multivariate analysis. Jpn. J. Zootech. Sci. 1988, 59, 952–960. (In Japanese) [Google Scholar] [CrossRef]
- Nomura, T.; Honda, T.; Mukai, F. Inbreeding and effective population size of Japanese Black cattle. J. Anim. Sci. 2001, 79, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Nomura, T.; Fukushima, M.; Mukai, F. Genetic diversity of a closed population of Japanese Black cattle in Hyogo prefecture. Anim. Sci. J. 2011, 72, 378–385. [Google Scholar] [CrossRef]
- Nishimura, S.; Watanabe, T.; Mizoshita, K.; Tatsuda, K.; Fujita, T.; Watanabe, N.; Sugimoto, Y.; Takasuga, A. Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012, 13, 40. [Google Scholar] [CrossRef] [Green Version]
- Setoguchi, K.; Furuta, M.; Hirano, T.; Nagao, T.; Watanabe, T.; Sugimoto, Y.; Takasuga, A. Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-Met substitution in NCAPG as a positional candidate. BMC Genet. 2009, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Takasuga, A.; Sato, K.; Nakamura, R.; Saito, Y.; Sasaki, S.; Tsuji, T.; Suzuki, A.; Kobayashi, H.; Matsuhashi, T.; Setoguchi, K.; et al. Non-synonymous FGD3 variant as positional candidate for disproportional tall stature accounting for a carcass weight QTL (CW-3) and skeletal dysplasia in Japanese Black cattle. PLoS Genet. 2015, 11, e1005433. [Google Scholar] [CrossRef]
- Zoda, A.; Ogawa, S.; Matsuda, H.; Taniguchi, Y.; Watanabe, T.; Sugimoto, Y.; Iwaisaki, H. Homozygosity region analysis using commercial single nucleotide polymorphism markers in Japanese Black cattle population. J. Anim. Genet. 2022, in press.
- Ookura, K.; Akiyama, T.; Yoshida, E.; Fukushima, M.; Iwamoto, E.; Oka, A.; Matsumoto, H.; Sasazaki, S.; Oyama, K.; Mannen, H. Effects of genes on economically important traits of Japanese Black cattle in Hyogo population. Nihon Chikusan Gakkaiho 2013, 84, 157–162. (In Japanese) [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Lund, M.S.; Wang, Y.; Su, G. Genomic predictions across Nordic Holstein and Nordic Red using the genomic best linear unbiased prediction model with different genomic relationship matrices. J. Anim. Breed. Genet. 2014, 131, 249–257. [Google Scholar] [CrossRef]
- Wientjes, Y.C.J.; Bijma, P.; Vandenplas, J.; Calus, M.P.L. Multi-population genomic relationships for estimating current genetic variances within and genetic correlations between populations. Genetics 2017, 207, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Doekes, H.P.; Veerkamp, R.F.; Bijma, P.; Hiemstra, S.J.; Windig, J.J. Trends in genome-wide and region-specific genetic diversity in the Dutch-Flemish Holstein-Friesian breeding program from 1986 to 2015. Genet. Sel. Evol. 2018, 50, 15. [Google Scholar] [CrossRef]
- Forutan, M.; Ansari Mahyari, S.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doublet, A.C.; Croiseau, P.; Fritz, S.; Michenet, A.; Hozé, C.; Danchin-Burge, C.; Laloë, D.; Restoux, G. The impact of genomic selection on genetic diversity and genetic gain in three French dairy cattle breeds. Genet. Sel. Evol. 2019, 51, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozada-Soto, E.A.; Maltecca, C.; Lu, D.; Miller, S.; Cole, J.B.; Tiezzi, F. Trends in genetic diversity and the effect of inbreeding in American Angus cattle under genomic selection. Genet. Sel. Evol. 2021, 53, 50. [Google Scholar] [CrossRef]
- Scott, B.A.; Haile-Mariam, M.; Cocks, B.G.; Pryce, J.E. How genomic selection has increased rates of genetic gain and inbreeding in the Australian national herd, genomic information nucleus, and bulls. J. Dairy Sci. 2021, 104, 11832–11849. [Google Scholar] [CrossRef]
- Ablondi, M.; Sabbioni, A.; Stocco, G.; Cipolat-Gotet, C.; Dadousis, C.; van Kaam, J.T.; Finocchiaro, R.; Summer, A. Genetic diversity in the Italian Holstein dairy cattle based on pedigree and SNP data prior and after genomic selection. Front. Vet. Sci. 2022, 8, 773985. [Google Scholar] [CrossRef]
- Lozada-Soto, E.A.; Tiezzi, F.; Jiang, J.; Cole, J.B.; VanRaden, P.M.; Maltecca, C. Genomic characterization of autozygosity and recent inbreeding trends in all major breeds of US dairy cattle. J. Dairy Sci. 2022, 105, 8956–8971. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, E.C.; Edel, C.; Emmerling, R.; Götz, K.U. How imputation errors bias genomic predictions. J. Dairy Sci. 2015, 98, 4131–4138. [Google Scholar] [CrossRef] [Green Version]
- Dadousis, C.; Ablondi, M.; Cipolat-Gotet, C.; van Kaam, J.T.; Marusi, M.; Cassandro, M.; Sabbioni, A.; Summer, A. Genomic inbreeding coefficients using imputed genotypes: Assessing different estimators in Holstein-Friesian dairy cows. J. Dairy Sci. 2022, 105, 5926–5945. [Google Scholar] [CrossRef]
- Ogawa, S.; Matsuda, H.; Taniguchi, Y.; Watanabe, T.; Takasuga, A.; Sugimoto, Y.; Iwaisaki, H. Accuracy of imputation of single nucleotide polymorphism marker genotypes from low-density panels in Japanese Black cattle. Anim. Sci. J. 2016, 87, 3–12. [Google Scholar] [CrossRef]
- Patry, C.; Ducrocq, V. Evidence of biases in genetic evaluations due to genomic preselection in dairy cattle. J. Dairy Sci. 2011, 94, 1011–1020. [Google Scholar] [CrossRef]
- Vitezica, Z.G.; Aguilar, I.; Misztal, I.; Legarra, A. Bias in genomic predictions for populations under selection. Genet. Res. 2011, 93, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Blasco, A.; Toro, M.A. A short critical history of the application of genomics to animal breeding. Livest. Sci. 2014, 166, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Heidaritabar, M.; Vereijken, A.; Muir, W.M.; Meuwissen, T.; Cheng, H.; Megens, H.J.; Groenen, M.A.; Bastiaansen, J.W. Systematic differences in the response of genetic variation to pedigree and genome-based selection methods. Heredity 2014, 113, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Sørensen, A.C.; Meuwissen, T.H.; Berg, P. Allele frequency changes due to hitch-hiking in genomic selection programs. Genet. Sel. Evol. 2014, 46, 8. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Romano, F.; Villanueva, B.; Fernández, J.; Woolliams, J.A.; Pong-Wong, R. The use of genomic coancestry matrices in the optimisation of contributions to maintain genetic diversity at specific regions of the genome. Genet. Sel. Evol. 2016, 48, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, B.J.; Pryce, J.; Chamberlain, A.J.; Bowman, P.J.; Goddard, M.E. Genetic architecture of complex traits and accuracy of genomic prediction: Coat colour, milk-fat percentage, and type in Holstein cattle as contrasting model traits. PLoS Genet. 2010, 6, e1001139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, S.M.; Thomsen, B.; Madsen, P.; Sørensen, P. Partitioning of genomic variance reveals biological pathways associated with udder health and milk production traits in dairy cattle. Genet. Sel. Evol. 2015, 47, 60. [Google Scholar] [CrossRef]




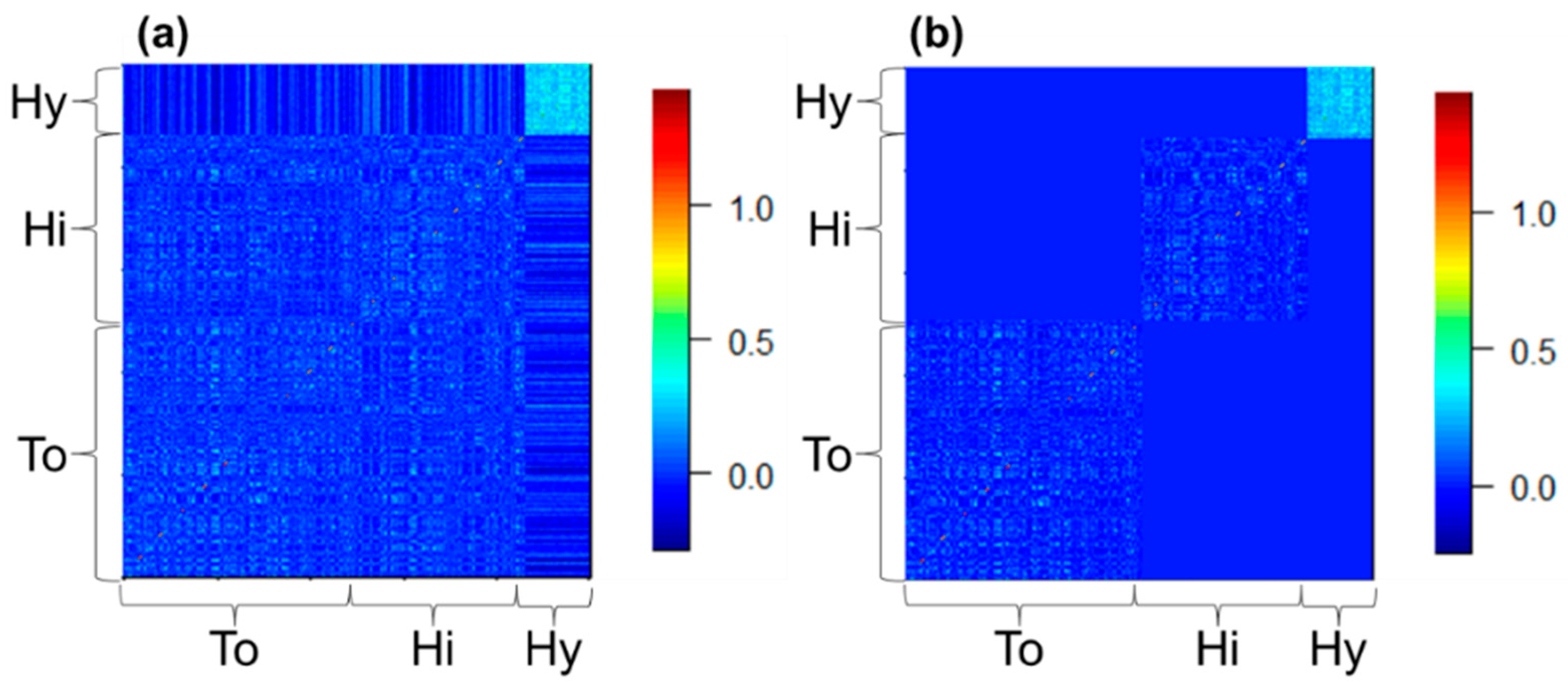{kind=link}
{kind=link}
{kind=link}
| Item; Unit | Tottori | Hiroshima | Hyogo | ||||||
|---|---|---|---|---|---|---|---|---|---|
| N | Mean | SD | N | Mean | SD | N | Mean | SD | |
| Age at slaughter; month | 1036 | 28.9 | 1.2 | 733 | 30.0 | 2.1 | 279 | 31.0 | 1.1 |
| Cold carcass weight; kg | 474.2 | 50.2 | 485.4 | 57.1 | 408.2 | 39.4 | |||
| Marbling score; 1 (null) to 12 (very abundance) | 5.8 | 2.0 | 4.1 | 1.4 | 6.5 | 1.9 | |||
| Model | DIC | Heritability | Genetic Correlation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Value | SE | Value | SE | Value | SE | Value | SE | Value | SE | Value | SE | ||
| Cold carcass weight | |||||||||||||
| 1 | 0 | 1163.6 | 116.2 | - | - | 1217.7 | 72.6 | 2381.3 | 89.3 | 0.49 | 0.04 | - | - |
| 2 | −25.4 | 748.2 | 166.2 | 485.2 | 156.4 | 1173.9 | 74.8 | 2407.4 | 90.8 | 0.51 | 0.04 | 0.61 | 0.12 |
| 3 | −14.0 | - | - | 1211.4 | 123.6 | 1183.5 | 76.9 | 2394.9 | 90.0 | 0.51 | 0.04 | - | - |
| 4 | −101.1 | 941.1 | 126.9 | 390.9 | 105.3 | 1081.9 | 79.8 | 2413.8 | 90.5 | 0.55 | 0.04 | 0.71 | 0.07 |
| 5 | −91.5 | - | - | 1375.2 | 137.9 | 1057.9 | 88.5 | 2433.0 | 90.3 | 0.56 | 0.04 | - | - |
| Marbling score | |||||||||||||
| 1 | 0 | 1.26 | 0.15 | - | - | 1.90 | 0.10 | 3.16 | 0.11 | 0.40 | 0.04 | - | - |
| 2 | −18.8 | 0.72 | 0.18 | 0.63 | 0.17 | 1.84 | 0.10 | 3.19 | 0.12 | 0.42 | 0.04 | 0.53 | 0.12 |
| 3 | −2.4 | - | - | 1.28 | 0.15 | 1.88 | 0.11 | 3.15 | 0.11 | 0.40 | 0.04 | - | - |
| 4 | −47.0 | 0.98 | 0.16 | 0.43 | 0.11 | 1.76 | 0.11 | 3.18 | 0.11 | 0.44 | 0.04 | 0.69 | 0.08 |
| 5 | 42.4 | - | - | 1.23 | 0.15 | 1.89 | 0.12 | 3.13 | 0.11 | 0.39 | 0.04 | - | - |
| Model | Effect | Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | C | To-Hi | Hy | To-Hi | Hy | C | To | Hi | Hy | To | Hi | Hy | ||
| 1 | C | 1.00 | 0.95 | 0.27 | 0.96 | 0.31 | 1.00 | 0.66 | 0.61 | 0.26 | 0.73 | 0.67 | 0.31 | |
| 2 | C | 1.00 | 0.95 | 0.29 | 0.96 | 0.33 | 1.00 | 0.65 | 0.61 | 0.27 | 0.73 | 0.67 | 0.33 | |
| To-Hi | 0.91 | 0.91 | −0.02 | 1.00 | 0.02 | 0.95 | 0.69 | 0.64 | −0.05 | 0.75 | 0.70 | 0.02 | ||
| Hy | 0.39 | 0.40 | −0.03 | 0.02 | 0.98 | 0.28 | −0.04 | −0.02 | 1.00 | 0.02 | 0.02 | 0.98 | ||
| 3 | To-Hi | 0.92 | 0.92 | 1.00 | 0.03 | 0.05 | 0.96 | 0.69 | 0.64 | 0.00 | 0.75 | 0.70 | 0.07 | |
| Hy | 0.41 | 0.42 | 0.01 | 0.99 | 0.07 | 0.32 | −0.01 | 0.01 | 0.96 | 0.06 | 0.06 | 1.00 | ||
| 4 | C | 0.99 | 1.00 | 0.91 | 0.39 | 0.92 | 0.41 | 0.65 | 0.62 | 0.27 | 0.74 | 0.68 | 0.32 | |
| To | 0.77 | 0.77 | 0.84 | 0.00 | 0.84 | 0.02 | 0.78 | −0.09 | −0.01 | 0.97 | 0.01 | −0.01 | ||
| Hi | 0.40 | 0.39 | 0.46 | −0.07 | 0.45 | −0.06 | 0.40 | −0.07 | −0.08 | 0.03 | 0.97 | 0.01 | ||
| Hy | 0.38 | 0.39 | −0.03 | 1.00 | 0.01 | 0.99 | 0.38 | −0.05 | −0.04 | 0.00 | 0.00 | 0.96 | ||
| 5 | To | 0.81 | 0.81 | 0.86 | 0.04 | 0.87 | 0.06 | 0.81 | 0.99 | −0.02 | 0.03 | 0.13 | 0.06 | |
| Hi | 0.52 | 0.52 | 0.57 | 0.00 | 0.57 | 0.02 | 0.53 | 0.10 | 0.92 | −0.01 | 0.16 | 0.01 | ||
| Hy | 0.41 | 0.42 | 0.01 | 0.99 | 0.05 | 1.00 | 0.41 | 0.02 | −0.06 | 0.99 | 0.06 | 0.05 | ||
| Prefecture | Cold Carcass Weight | Marbling Score | ||||||
|---|---|---|---|---|---|---|---|---|
| Model 2 | Model 3 | Model 4 | Model 5 | Model 2 | Model 3 | Model 4 | Model 5 | |
| Pearson correlation | ||||||||
| Tottori | 0.999 | 0.996 | 0.992 | 0.968 | 0.998 | 0.993 | 0.995 | 0.976 |
| Hiroshima | 0.999 | 0.997 | 0.991 | 0.965 | 0.997 | 0.990 | 0.977 | 0.896 |
| Hyogo | 0.982 | 0.914 | 0.987 | 0.912 | 0.988 | 0.957 | 0.994 | 0.957 |
| Spearman’s rank correlation | ||||||||
| Tottori | 0.999 | 0.996 | 0.992 | 0.968 | 0.998 | 0.993 | 0.995 | 0.976 |
| Hiroshima | 0.999 | 0.997 | 0.990 | 0.961 | 0.997 | 0.990 | 0.977 | 0.892 |
| Hyogo | 0.980 | 0.912 | 0.985 | 0.910 | 0.987 | 0.957 | 0.993 | 0.957 |
| Model | Tottori | Hiroshima | Hyogo | Mean + Effect of Prefecture | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Tottori | Hiroshima | Hyogo | |
| Cold carcass weight | |||||||||
| 1 | 0 | 29.6 | −10.1 | 32.2 | −68.9 | 18.2 | 0 | 7.2 | −102.7 |
| 2 | 0 | 30.2 | −9.8 | 32.9 | −42.7 | 18.4 | 0 | 7.1 | −102.1 |
| 3 | 0 | 29.9 | −9.4 | 32.5 | −5.8 | 17.3 | 0 | 7.7 | −102.2 |
| 4 | 0 | 31.3 | −10.8 | 34.2 | −55.3 | 19.6 | 0 | 7.7 | −98.8 |
| 5 | 0 | 31.1 | −10.8 | 33.8 | −5.9 | 19.0 | 0 | 9.2 | −96.9 |
| Marbling score | |||||||||
| 1 | 0 | 0.93 | −0.36 | 0.76 | −0.08 | 0.78 | 0 | −1.90 | 0.59 |
| 2 | 0 | 0.96 | −0.36 | 0.78 | 0.33 | 0.83 | 0 | −1.92 | 0.61 |
| 3 | 0 | 0.93 | −0.35 | 0.76 | 0.60 | 0.79 | 0 | −1.90 | 0.59 |
| 4 | 0 | 1.03 | −0.39 | 0.72 | 0.15 | 0.86 | 0 | −1.88 | 0.72 |
| 5 | 0 | 0.94 | −0.34 | 0.59 | 0.54 | 0.78 | 0 | −1.82 | 0.71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogawa, S.; Taniguchi, Y.; Watanabe, T.; Iwaisaki, H. Fitting Genomic Prediction Models with Different Marker Effects among Prefectures to Carcass Traits in Japanese Black Cattle. Genes 2023, 14, 24. https://doi.org/10.3390/genes14010024
Ogawa S, Taniguchi Y, Watanabe T, Iwaisaki H. Fitting Genomic Prediction Models with Different Marker Effects among Prefectures to Carcass Traits in Japanese Black Cattle. Genes. 2023; 14(1):24. https://doi.org/10.3390/genes14010024
Chicago/Turabian StyleOgawa, Shinichiro, Yukio Taniguchi, Toshio Watanabe, and Hiroaki Iwaisaki. 2023. "Fitting Genomic Prediction Models with Different Marker Effects among Prefectures to Carcass Traits in Japanese Black Cattle" Genes 14, no. 1: 24. https://doi.org/10.3390/genes14010024
APA StyleOgawa, S., Taniguchi, Y., Watanabe, T., & Iwaisaki, H. (2023). Fitting Genomic Prediction Models with Different Marker Effects among Prefectures to Carcass Traits in Japanese Black Cattle. Genes, 14(1), 24. https://doi.org/10.3390/genes14010024