

DelInsCaller: An Efficient Algorithm for Identifying Delins and Estimating Haplotypes from Long Reads with High Level of Sequencing Errors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
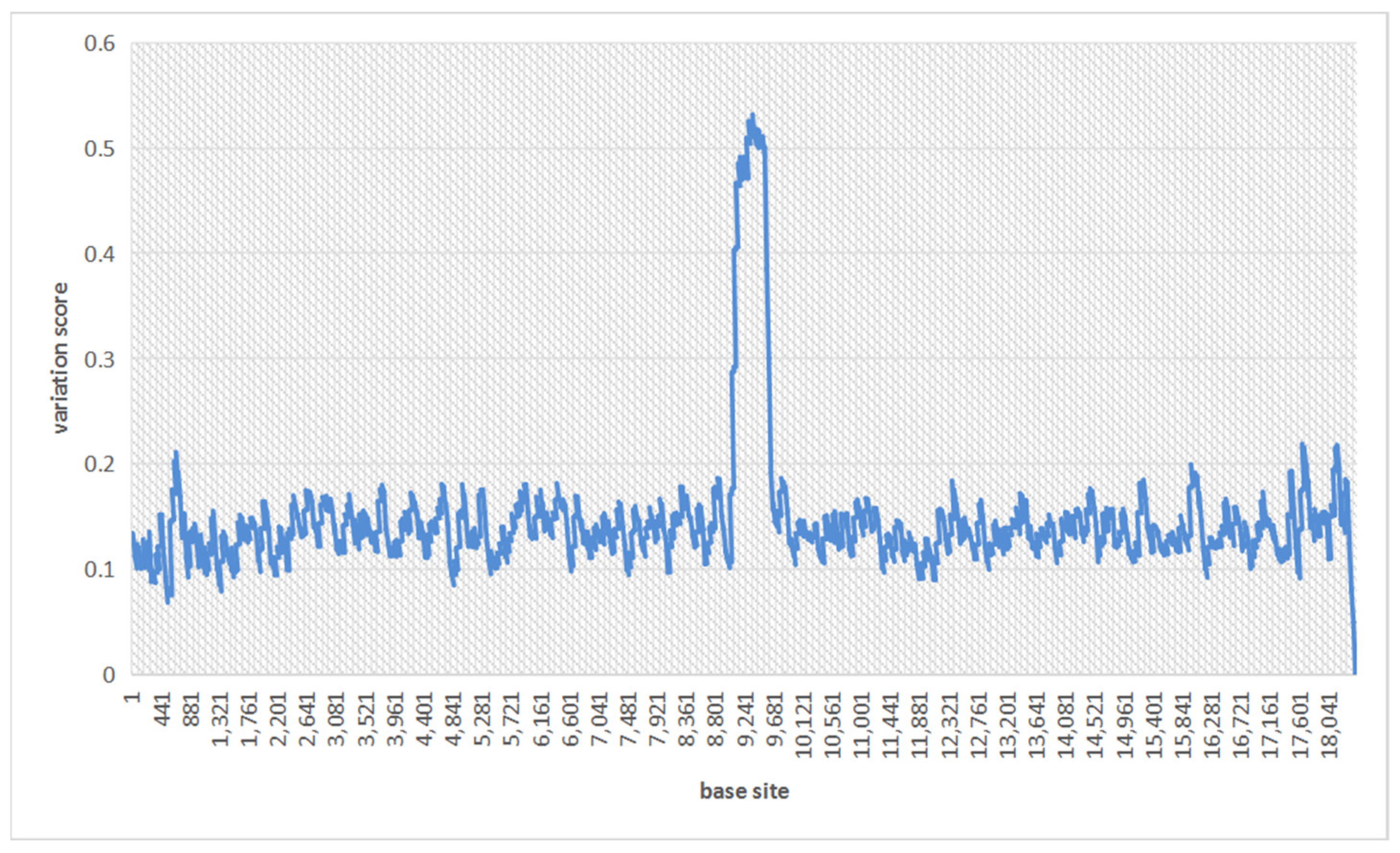
2.1. Identifying Regions of Variations
2.2. Classification of Variations
2.3. Locating the Start and End Sites of the Variation
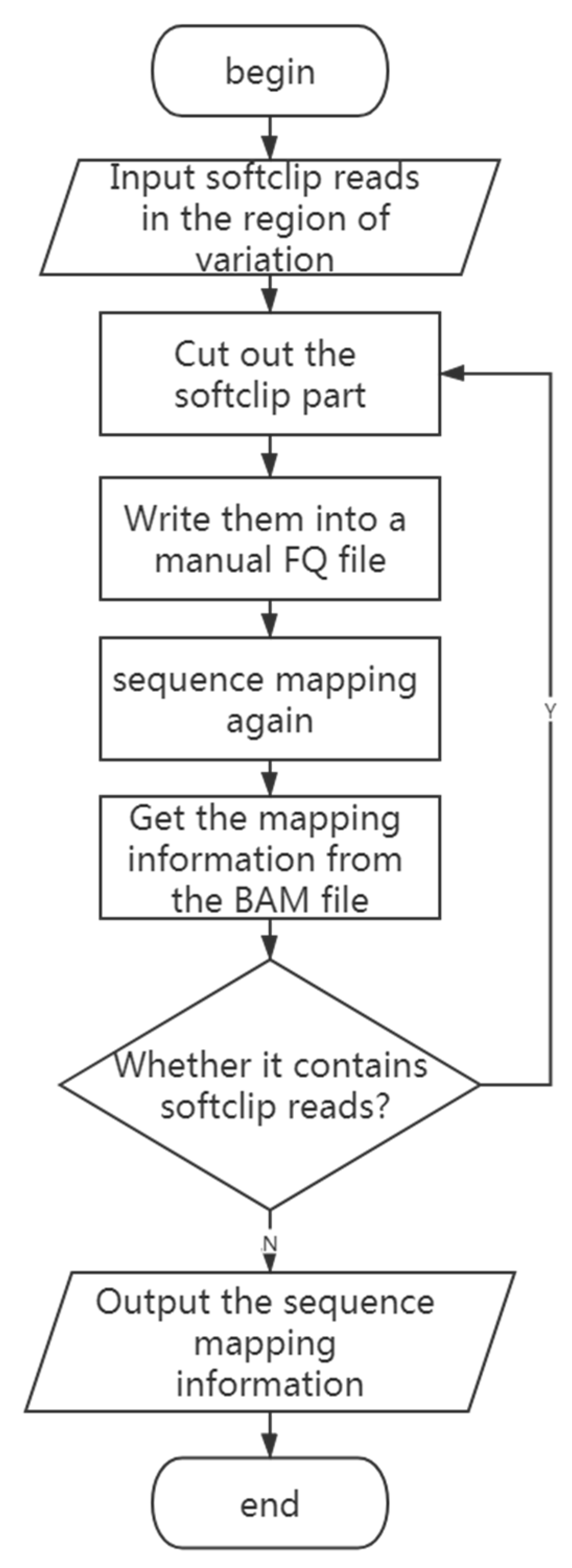
2.4. Finding the Source of the Inserted Fragment
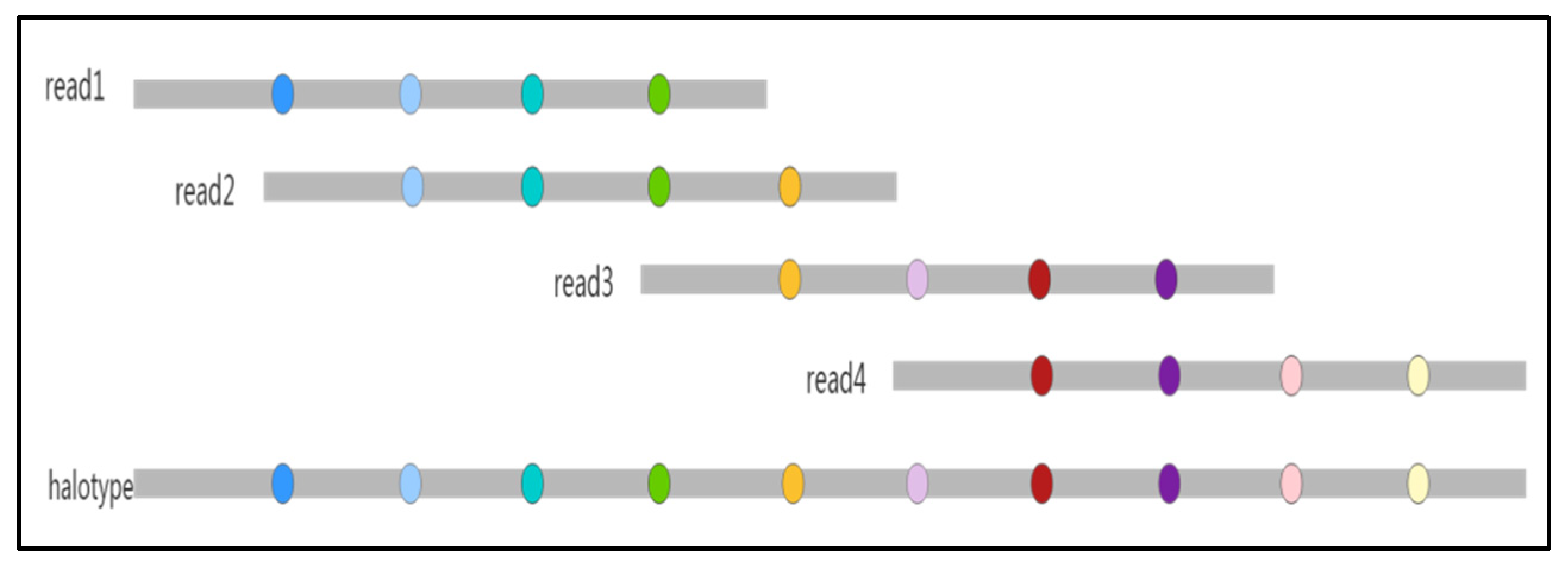
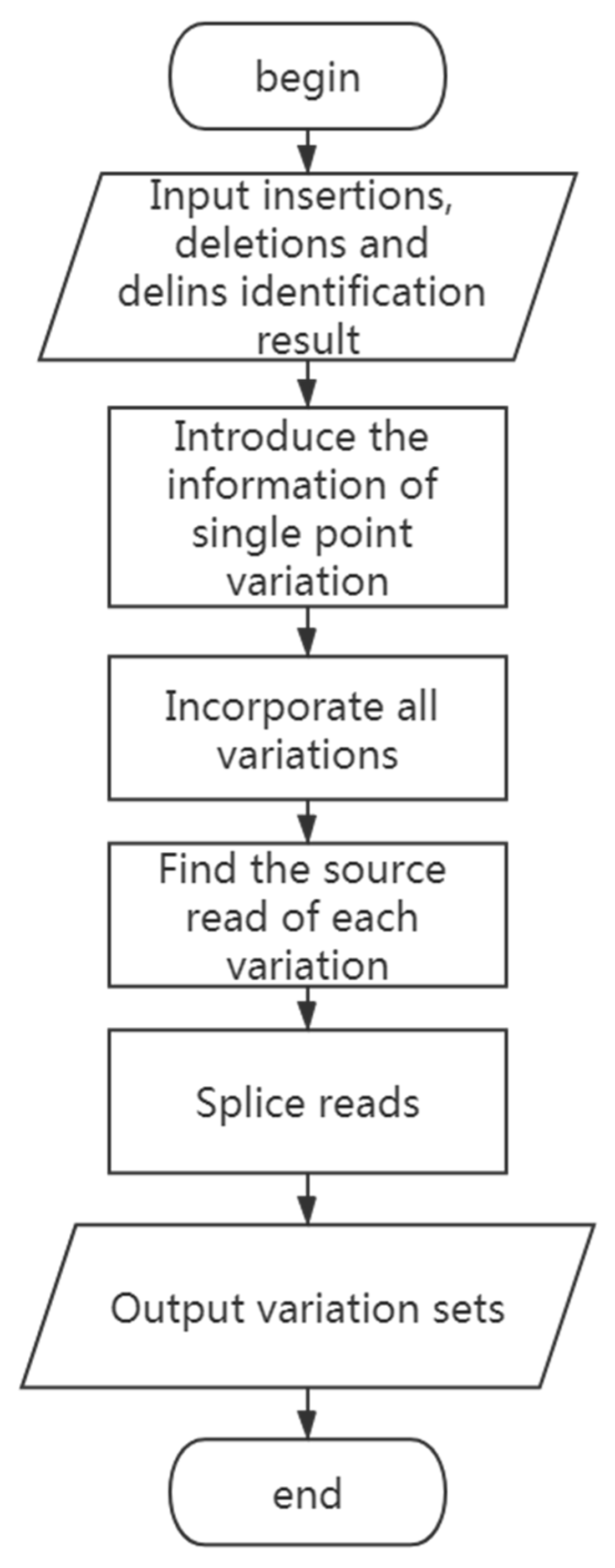
2.5. Estimating Haplotypes
2.6. Experimental Methods
2.6.1. Simulation Data Sets Experiments
2.6.2. Independent Validation Data Sets Experiments
3. Results and Discussion
3.1. Experiments under Different Data Characteristics
3.1.1. Experimental Results under Different Sequencing Depths
3.1.2. Experimental Results under Different Lengths of Delins
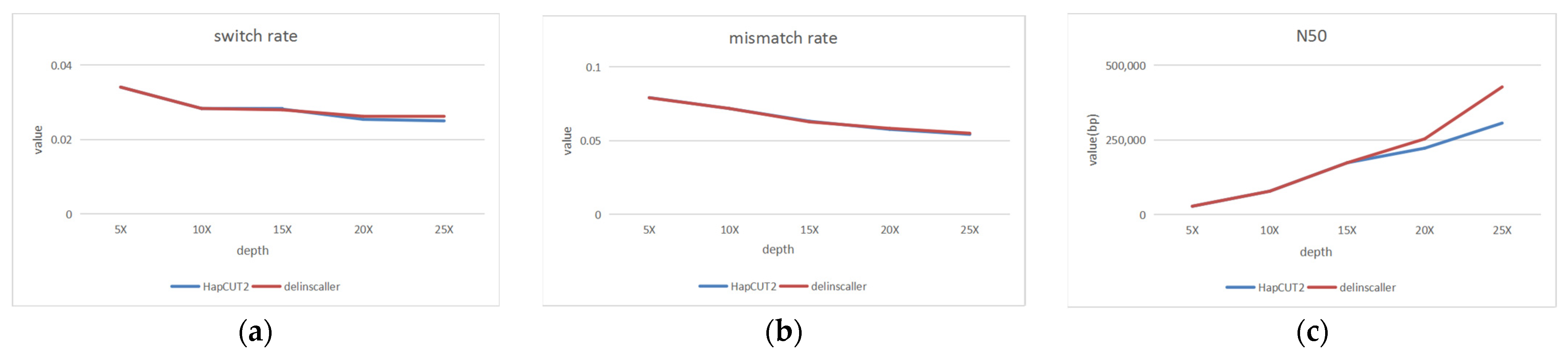
3.2. Haplotype Phasing Experiments
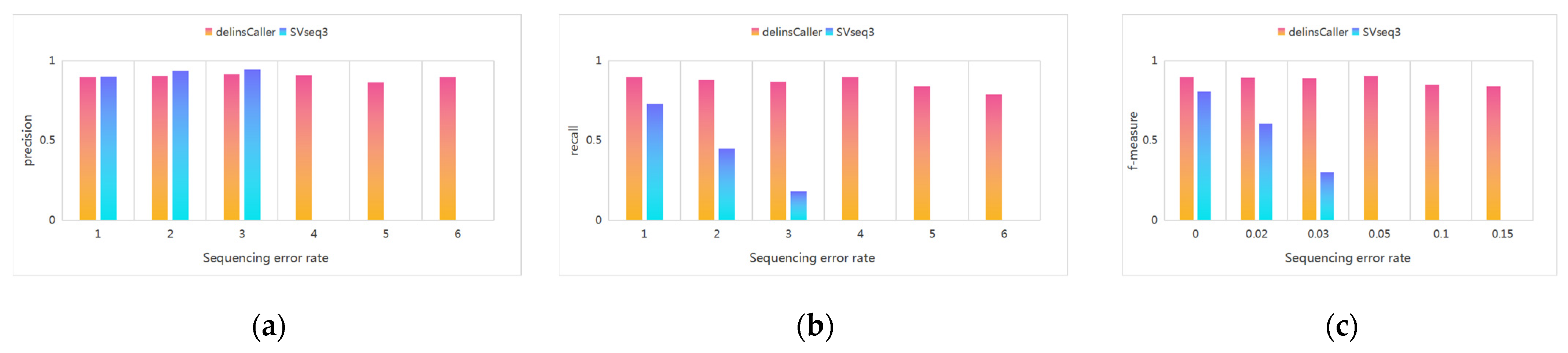
3.3. Comparison Experiment with SVseq3
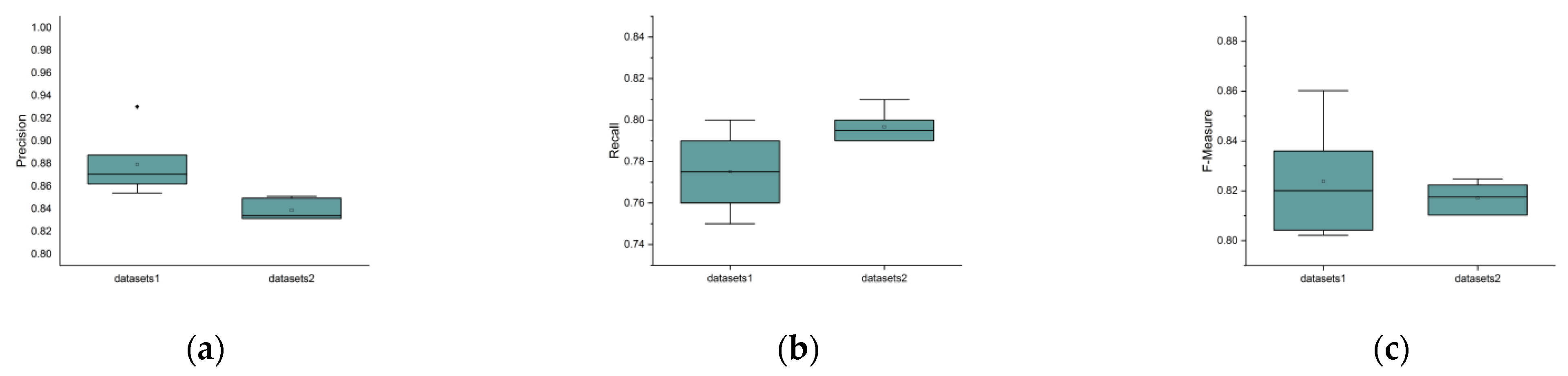
3.4. Independent Validation Data Sets Experiments
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Quinlan, A.R.; Hall, I.M. Characterizing complex structural variation in germline and somatic genomes. Trends Genet. 2012, 28, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, R.L.; Brand, H.; Redin, C.E.; Hanscom, C.; Antolik, C.; Stone, M.R.; Glessner, J.T.; Mason, T.; Pregno, G.; Dorrani, N.; et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. 2017, 18, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, K.; Wang, J.; Jayasinghe, R.; Lameijer, E.-W.; McMichael, J.F.; Ning, J.; McLellan, M.D.; Xie, M.; Cao, S.; Yellapantula, V.; et al. Systematic discovery of complex insertions and deletions in human cancers. Nat. Med. 2016, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef] [Green Version]
- Roerink, S.F.; van Schendel, R.; Tijsterman, M. Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res. 2014, 24, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Koole, W.; Van, S.R.; Karambelas, A.E.; van Heteren, J.T.; Okihara, K.L.; Tijsterman, M. A polymerase theta-dependent repair pathway suppresses extensive genomic instability at endogenous g4 DNA sites. Nat. Commun. 2014, 5, 3216. [Google Scholar] [CrossRef] [Green Version]
- Kwong, A.; Shin, V.Y.; Au, C.H.; Law, F.B.; Ho, D.N.; Ip, B.K.; Wong, A.T.; Lau, S.S.; To, R.M.; Choy, G.; et al. Detection of Germline Mutation in Hereditary Breast and/or Ovarian Cancers by Next-Generation Sequencing on a Four-Gene Panel. J. Mol. Diagn. 2016, 18, 580–594. [Google Scholar] [CrossRef] [Green Version]
- Garcia, C.; Lyon, L.; Littell, R.D.; Powell, C.B. Comparison of risk management strategies between women testing positive for a BRCA variant of unknown significance and women with known BRCA deleterious mutations. Genet. Med. 2014, 16, 896–902. [Google Scholar] [CrossRef] [Green Version]
- Kloosterman, W.P.; Francioli, L.C.; Hormozdiari, F.; Marschall, T.; Hehirkwa, J.Y.; Abdellaoui, A.; Lameijer, E.-W.; Moed, M.H.; Koval, V.; Renkens, I.; et al. Characteristics of de novo structural changes in the human genome. Genome Res. 2015, 25, 792. [Google Scholar] [CrossRef] [Green Version]
- Zheng, T.; Li, Y.; Geng, Y.; Zhao, Z.; Zhang, X.; Xiao, X.; Wang, J. CIGenotyper: A Machine Learning Approach for Genotyping Complex Indel Calls. Bioinform. Biomed. Eng. 2018, 10813, 473–485. [Google Scholar]
- Iakovishina, D.; Janoueix-Lerosey, I.; Barillot, E.; Regnier, M.; Boeva, V. SV-Bay: Structural variant detection in cancer genomes using a Bayesian approach with correction for GC-content and read mappability. Bioinformatics 2016, 32, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Au, C.H.; Leung, A.Y.H.; Kwong, A.; Chan, T.L.; Ma, E.S.K. INDELseek: Detection of complex insertions and deletions from next-generation sequencing data. BMC Genom. 2017, 18, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaisson, M.J.P.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2014, 517, 608–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Schatz, M.C. Genomic dark matter: The reliability of short read mapping illustrated by the genome mappability score. Bioinformatics 2012, 28, 2097–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2010, 472, 431–455. [Google Scholar] [CrossRef]
- John, H.; Chaisson, M.J.P.; Steinberg, K.M. Corrigendum Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2017, 27, 677–685. [Google Scholar]
- Zhang, X.; Chen, H.; Zhang, R.; Pei, J.; Wang, Y.; Zhao, Z.; Huang, Y.; Wang, J. Detecting complex indels with wide length-spectrum from the third generation sequencing data. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine, Kansas City, MO, USA, 13–16 November 2017; IEEE: Piscataway Township, NJ, USA, 2017; pp. 1980–1987. [Google Scholar]
- Chaisson, M.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef] [Green Version]
- Yukiteru, O.; Kiyoshi, A.; Michiaki, H. PBSIM: PacBio reads simulator—Toward accurate genome assembly. Bioinformatics 2013, 29, 119–121. [Google Scholar]
- Jiao, X.D.; He, X.; Qin, B.D.; Liu, K.; Wu, Y.; Liu, J.; Hou, T.; Zang, Y.S. The prognostic value of tumor mutation burden in EGFR-mutant advanced lung adenocarcinoma, an analysis based on cBioPortal data base. J. Thorac. Dis. 2019, 11, 4507–4515. [Google Scholar] [CrossRef]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Morton, N.E.; Zhang, W.; Taillon-Miller, P.; Ennis, S.; Kwok, P.Y.; Collins, A. The optimal measure of allelic association. Proc. Natl. Acad. Sci. USA 2001, 98, 5217–5221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahsan, M.U.; Liu, Q.; Fang, L.; Wang, K. NanoCaller for accurate detection of SNPs and indels in difficult-to-map regions from long-read sequencing by haplotype-aware deep neural networks. Genome Biol. 2021, 22, 261. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Zhang, X.; Qiang, G.; Wang, J. DelInsCaller: An Efficient Algorithm for Identifying Delins and Estimating Haplotypes from Long Reads with High Level of Sequencing Errors. Genes 2023, 14, 4. https://doi.org/10.3390/genes14010004
Wang S, Zhang X, Qiang G, Wang J. DelInsCaller: An Efficient Algorithm for Identifying Delins and Estimating Haplotypes from Long Reads with High Level of Sequencing Errors. Genes. 2023; 14(1):4. https://doi.org/10.3390/genes14010004
Chicago/Turabian StyleWang, Shenjie, Xuanping Zhang, Geng Qiang, and Jiayin Wang. 2023. "DelInsCaller: An Efficient Algorithm for Identifying Delins and Estimating Haplotypes from Long Reads with High Level of Sequencing Errors" Genes 14, no. 1: 4. https://doi.org/10.3390/genes14010004
APA StyleWang, S., Zhang, X., Qiang, G., & Wang, J. (2023). DelInsCaller: An Efficient Algorithm for Identifying Delins and Estimating Haplotypes from Long Reads with High Level of Sequencing Errors. Genes, 14(1), 4. https://doi.org/10.3390/genes14010004