
Exome Array Analysis of 9721 Ischemic Stroke Cases from the SiGN Consortium

 , , , , ,
, , , , ,  , ,
on behalf of the SiGN Consortiumadd
Show full author list
, ,
on behalf of the SiGN Consortiumadd
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Genotyping
2.2. Genotype Quality Control
2.3. Population Structure Analysis Using Admixture and PCA
2.4. Association Analysis
2.5. Replication
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 8 August 2022).
- Malik, R.; Chauhan, G.; Traylor, M.; Sargurupremraj, M.; Okada, Y.; Mishra, A.; Rutten-Jacobs, L.; Giese, A.K.; van der Laan, S.W.; Gretarsdottir, S.; et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018, 50, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Malik, R.; Hachiya, T.; Jürgenson, T.; Namba, S.; Posner, D.C.; Kamanu, F.K.; Koido, M.; Le Grand, Q.; Shi, M.; et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 2022, 611, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Jaworek, T.; Ryan, K.A.; Gaynor, B.J.; McArdle, P.F.; Stine, O.C.; TD, O.C.; Lopez, H.; Aparicio, H.J.; Gao, Y.; Lin, X.; et al. Exome array analysis of early-onset ischemic stroke. Stroke J. Cereb. Circ. 2020, 51, 3356–3360. [Google Scholar] [CrossRef] [PubMed]
- Auer, P.L.; Nalls, M.; Meschia, J.F.; Worrall, B.B.; Longstreth, W.T., Jr.; Seshadri, S.; Kooperberg, C.; Burger, K.M.; Carlson, C.S.; Carty, C.L.; et al. Rare and coding region genetic variants associated with risk of ischemic stroke: The NHLBI Exome Sequence Project. JAMA Neurol. 2015, 72, 781–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NINDS Stroke Genetics Network; International Stroke Genetics Consortium. Loci associated with ischaemic stroke and its subtypes (SiGN): A genome-wide association study. Lancet Neurol. 2016, 15, 174–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnega, A.; Faul, J.D.; Ofstedal, M.B.; Langa, K.M.; Phillips, J.W.; Weir, D.R. Cohort Profile: The Health and Retirement Study (HRS). Int. J. Epidemiol. 2014, 43, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Z.; Absher, D.M.; Tang, H.; Southwick, A.M.; Casto, A.M.; Ramachandran, S.; Cann, H.M.; Barsh, G.S.; Feldman, M.; Cavalli-Sforza, L.L.; et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008, 319, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Nielsen, J.B.; Fritsche, L.G.; Dey, R.; Gabrielsen, M.E.; Wolford, B.N.; LeFaive, J.; VandeHaar, P.; Gagliano, S.A.; Gifford, A.; et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018, 50, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Haessler, J.W.; Manansala, R.; Wiggins, K.L.; Moscati, A.; Beiser, A.; Heard-Costa, N.L.; Sarnowski, C.; Raffield, L.M.; Chung, J.; et al. Whole-genome sequencing association analyses of stroke and its subtypes in ancestrally diverse populations from Trans-Omics for Precision Medicine Project. Stroke J. Cereb. Circ. 2022, 53, 875–885. [Google Scholar] [CrossRef] [PubMed]
- UK Biobank Outcome Adjudication Group. UK Biobank Algorithmically-Derived Outcomes (ADOs), Version 2. Available online: https://biobank.ndph.ox.ac.uk/showcase/ukb/docs/alg_outcome_main.pdf (accessed on 23 August 2022).
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cases | Controls | |
|---|---|---|
| N | 9721 | 12,345 |
| Age (or Age of onset for cases) | 67.0 ± 14.0 | 57.0 ± 9.7 |
| Age range (yrs) | 14–104 | 17–94 |
| % Female | 4662 (73.4%) | 7335 (78.2%) |
| Self-reported ancestry | ||
| EUR | 7138 (73.4%) | 9659 (78.2%) |
| AFR | 1022 (10.5%) | 1358 (11.0%) |
| Hispanic | 893 (9.2%) | 1034 (8.4%) |
| Other | 666 (6.9%) | 0 |
| unspecified | 2 (0.2%) | 294 (2.4%) |
| Genetic ancestry (computed) | ||
| EUR | 7921 (81.5%) | 9911 (80.3%) |
| AFR | 1044 (10.7%) | 1353 (11.0%) |
| Other | 756 (7.8%) | 1081 (8.8%) |
| Number of subjects excluded in QC analysis based on unrelated subjects (but included in association analysis) | 246 (at pi-hat > 0.1875) | 0 |
| TOAST classification of IS | ||
| Cardioembolism | 2210 (10.0%) | |
| Large-Artery Atherosclerosis | 1297 (5.9%) | |
| Small-Artery Occlusion | 2218 (10.1%) | |
| Other Known Causes | 296 (1.34%) | |
| Undetermined | 1831 (8.3%) | |
| Unknown | 14,214 (64.4%) |
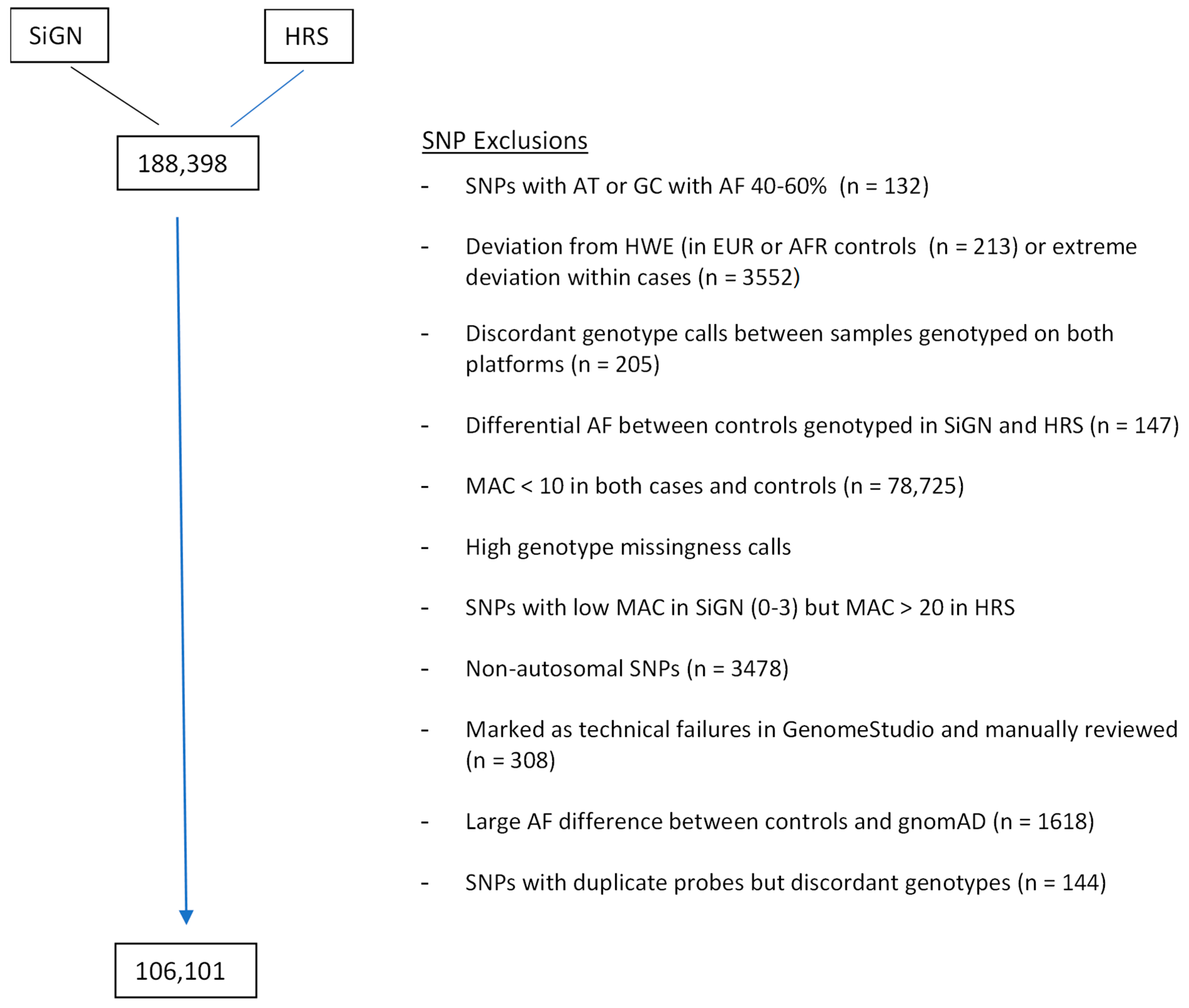
| 1. MERGE HRS + SiGN | 198,811 SNPs | |
|---|---|---|
| 2. Remove SNPs designed for technical purposes such as randomly selected synonymous variants as annotated by CHARGE (see Supplemental Materials) | 188,398 remained | |
| 3. SNP QC filter | Excluded SNPs * | |
| SNPs with genotype AT or GC if AF is between 40–60% | 132 | |
| deviation from HWE in HRS controls( p < 1.0 × 10−5 (EUR), p < 1.0 × 10−10 (AFR)) | 162 | |
| deviation from HWE in controls from SIGN ( p < 1.0 × 10−5 (EUR)) | 51 | |
| deviation from HWE in stroke cases (p < 1.0 × 10−20 (EUR), 1.0 × 10−10 (AFR)) | 3552 * | |
| discordant call > 0 among technical duplicated samples from 51 subjects genotyped on both platforms (29 HRS samples and 22 WUSTL samples) | 205 | |
| large allele frequency differences in EUR (EWAS p < 1.0 × 10−3 using the SAIGE method) between control samples genotyped in SiGN and HRS; the p < 1.0 × 10−3 cutoff was chosen through examining the outliers on the distribution plots of the resulting p-values. | 147 | |
| Missing rate >2.5% in SiGN or HRS for non-rare variants with MAF ≥ 1%, | 1145 | |
| Missing rate >0.8% in SiGN or HRS for rare variants with MAF < 1% | 43,289 | |
| Possibly undercalled in SiGN: MAC ≤ 3 in SiGN (all ancestries), but > 20 in HRS EURs | 66 | |
| Marked as technical failure in HRS upon additional manual review of Genome Studio clustering plots | 308 | |
| Allele frequency differ significantly between HRS EUR samples and gnomAD non-Finnish EUR (p < 1.0 × 10−5 for rare variants, p < 1.0 × 10−10 for low frequency and common variants) | 1618 | |
| duplicate probes on SIGN 5MplusExome array with discordant allele frequencies (Fisher exact p < 0.05) | 144 | |
| SNPs with low MAC (<10) | 78,725 | |
| Non-autosomal SNPs | 3478 | |
| Number of SNPs remaining | 106,101 | |
| 4. Both SNP-specific and study site specific masking for variants from SIGN samples | Affected SNPs | |
| variants showed substantial allele frequency differences between a particular site and the remaining samples (p < 1.0 × 10−3 and AC > 10) | 993 | |
| variants showed substantial call rate differences between a particular site and the remaining samples (differential missingness p value < 5.0 × 10−7) | 12,691 | |
| Number of SNPs remaining | 106,101 | |
| SNP | rs Number | Gene | Function | AA Change | A2 | A1 | OR | p-Value |
|---|---|---|---|---|---|---|---|---|
| exm1345082 | rs192153785 | GH2 | missense | Q228P | T | G | 7.45 | 1.92 × 10−8 |
| exm1501517 | rs140922537 | ZNF765 | missense | P270S | C | T | 6.99 | 5.63 × 10−10 |
| exm1562153 | rs143510517 | TPTE | missense | R274W | G | A | 6.56 | 7.17 × 10−12 |
| exm21949 | rs373898350 | NBPF1 | unknown | G | T | 6.73 | 3.56 × 10−7 | |
| exm365204 | rs141845742 | SPATA16 | stoploss | X570Q | A | G | 7.90 | 2.56 × 10−7 |
| exm384695 | rs149905649 | DOK7 | missense | R92W | C | T | 0.16 | 3.67 × 10−8 |
| exm552854 | rs62619974 | MEP1A | missense | K396R | A | G | 6.77 | 1.04 × 10−11 |
| exm558342 | rs199585353 | PRIM2 | unknown | G | T | 5.51 | 8.20 × 10−8 | |
| exm615057 | rs375144101 | TRGC1 | stoploss | Ter174LysextTer17 | A | T | 7.35 | 4.46 × 10−10 |
| exm791656 | rs142792732 | DDX31 | missense | E33K | C | T | 0.16 | 2.38 × 10−17 |
| exm90767 | rs372423248 | SEC22B | unknown | A | C | 7.41 | 2.57 × 10−8 | |
| exm90783 | rs373433490 | SEC22B | unknown | C | T | 6.74 | 1.65 × 10−10 | |
| exm913753 | rs79336999 | PATL1 | missense | Y645C | T | C | 7.52 | 2.10 × 10−13 |
| exmrs507666 | rs507666 | ABO | intronic | G | A | 1.17 | 4.79 × 10−8 | |
| exmrs635634 | rs635634 | ABO | Intergenic | C | T | 1.17 | 6.12 × 10−8 |
| Ancestry = ALL | EUROPEANS ONLY | AFRICAN AMERICANS ONLY | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CASES (n = 9721) | CONTROLS (n = 12,345) | CASES (n = 7138) | CONTROLS (n = 9659) | CASES (n = 1022) | CONTROLS (n = 1358) | |||||||||||||
| SNP | rs Nnumber | Gene | A2/A1 | MAF | AC | MAF | AC | MAF gnomAD (TOTAL) | MAF | AC | MAF | AC | MAF gnomAD (EUR Non-FINNISH) | MAF | AC | MAF | AC | MAF gnomAD (AFR/AFR AM) |
| exm1345082 | rs192153785 | GH2 | T/G | 0.21% | 40 | 0.00% | 0 | 0.15% | 0.01% | 2 | 0.00% | 0 | 0.00% | 1.58% | 33 | 0.00% | 0 | 1.44% |
| exm1501517 | rs140922537 | ZNF765 | C/T | 0.25% | 49 | 0.01% | 2 | 0.25% | 0.03% | 5 | 0.00% | 0 | 0.01% | 1.92% | 40 | 0.04% | 1 | 2.43% |
| exm1562153 | rs143510517 | TPTE | G/A | 0.36% | 69 | 0.01% | 2 | 0.07% | 0.04% | 6 | 0.00% | 0 | 0.00% | 2.55% | 53 | 0.04% | 1 | 0.77% |
| exm21949 | rs373898350 | NBPF1 | G/T | 0.18% | 35 | 0.00% | 1 | 0.00% | 0.01% | 1 | 0.00% | 0 | 0.00% | 1.35% | 28 | 0.04% | 1 | 0.03% |
| exm365204 | rs141845742 | SPATA16 | A/G | 0.16% | 31 | 0.00% | 0 | 0.12% | 0.00% | 0 | 0.00% | 0 | 0.00% | 1.29% | 27 | 0.00% | 0 | 1.17% |
| exm384695 | rs149905649 | DOK7 | C/T | 0.00% | 0 | 0.22% | 54 | 0.22% | 0.00% | 0 | 0.06% | 10 | 0.09% | 0.00% | 0 | 1.45% | 39 | 1.66% |
| exm552854 | rs62619974 | MEP1A | A/G | 0.34% | 67 | 0.00% | 1 | 0.36% | 0.01% | 1 | 0.00% | 0 | 0.01% | 2.83% | 59 | 0.00% | 0 | 3.08% |
| exm558342 | rs199585353 | PRIM2 | G/T | 0.21% | 40 | 0.02% | 6 | 0.02% | 0.25% | 40 | 0.00% | 0 | 0.04% | 0.00% | 0 | 0.00% | 0 | 0.01% |
| exm615057 | rs375144101 | TRGC1 | A/T | 0.26% | 50 | 0.00% | 1 | 0.13% | 0.04% | 6 | 0.00% | 0 | 0.01% | 1.63% | 34 | 0.04% | 1 | 1.27% |
| exm791656 | rs142792732 | DDX31 | C/T | 0.00% | 0 | 0.58% | 143 | 0.37% | 0.00% | 0 | 0.03% | 5 | 0.00% | 0.00% | 0 | 4.77% | 128 | 4.11% |
| exm90767 | rs372423248 | SEC22B | A/C | 0.21% | 40 | 0.00% | 0 | 0.03% | 0.01% | 1 | 0.00% | 0 | 0.00% | 1.58% | 33 | 0.00% | 0 | 0.28% |
| exm90783 | rs373433490 | SEC22B | C/T | 0.27% | 53 | 0.01% | 3 | 0.01% | 0.02% | 3 | 0.00% | 0 | 0.00% | 2.11% | 44 | 0.12% | 3 | 0.11% |
| exm913753 | rs79336999 | PATL1 | T/C | 0.37% | 71 | 0.00% | 0 | 0.22% | 0.00% | 0 | 0.00% | 0 | 0.00% | 3.21% | 67 | 0.00% | 0 | 2.41% |
| exm-rs507666 | rs507666 | ABO | G/A | 20.80% | 3589 | 17.99% | 4442 | 17.23% | 22.93% | 3207 | 19.14% | 3297 | 19.85% | 9.95% | 183 | 10.43% | 281 | 10.79% |
| exm-rs635634 | rs635634 | ABO | C/T | 20.78% | 3586 | 18.00% | 4443 | 17.17% | 22.94% | 3209 | 19.13% | 3296 | 19.79% | 9.68% | 178 | 10.50% | 283 | 10.75% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.; Nguyen, K.; Gaynor, B.J.; Ling, H.; Zhao, W.; McArdle, P.F.; O’Connor, T.D.; Stine, O.C.; Ryan, K.A.; Lynch, M.; et al. Exome Array Analysis of 9721 Ischemic Stroke Cases from the SiGN Consortium. Genes 2023, 14, 61. https://doi.org/10.3390/genes14010061
Xu H, Nguyen K, Gaynor BJ, Ling H, Zhao W, McArdle PF, O’Connor TD, Stine OC, Ryan KA, Lynch M, et al. Exome Array Analysis of 9721 Ischemic Stroke Cases from the SiGN Consortium. Genes. 2023; 14(1):61. https://doi.org/10.3390/genes14010061
Chicago/Turabian StyleXu, Huichun, Kevin Nguyen, Brady J. Gaynor, Hua Ling, Wei Zhao, Patrick F. McArdle, Timothy D. O’Connor, O. Colin Stine, Kathleen A. Ryan, Megan Lynch, and et al. 2023. "Exome Array Analysis of 9721 Ischemic Stroke Cases from the SiGN Consortium" Genes 14, no. 1: 61. https://doi.org/10.3390/genes14010061
APA StyleXu, H., Nguyen, K., Gaynor, B. J., Ling, H., Zhao, W., McArdle, P. F., O’Connor, T. D., Stine, O. C., Ryan, K. A., Lynch, M., Smith, J. A., Faul, J. D., Hu, Y., Haessler, J. W., Fornage, M., Kooperberg, C., on behalf of the Trans-Omics for Precision Medicine (TOPMed) Stroke Working Group, Perry, J. A., Hong, C. C., ... Mitchell, B. D., on behalf of the SiGN Consortium. (2023). Exome Array Analysis of 9721 Ischemic Stroke Cases from the SiGN Consortium. Genes, 14(1), 61. https://doi.org/10.3390/genes14010061