
Whole-Exome Sequencing of 24 Spanish Families: Candidate Genes for Non-Syndromic Pediatric Keratoconus

 , , , , , ,
, , , , , ,  and
and  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
1.1. Epidemiology and Etiology of Keratoconus
1.2. Pediatric Keratoconus
1.3. Diagnosis, Classification and Treatment of Keratoconus
1.4. Structure and Development of the Cornea and Its Histological Changes
1.4.1. Structure of the Cornea
1.4.2. Development of the Cornea
1.4.3. Histological Changes in Keratoconus
1.5. Pathophysiology of Keratoconus
1.5.1. Environmental Factors
1.5.2. Role of the Inflammatory Process
1.5.3. Role of Enzymes
1.5.4. Gender
1.5.5. Genetic Factors
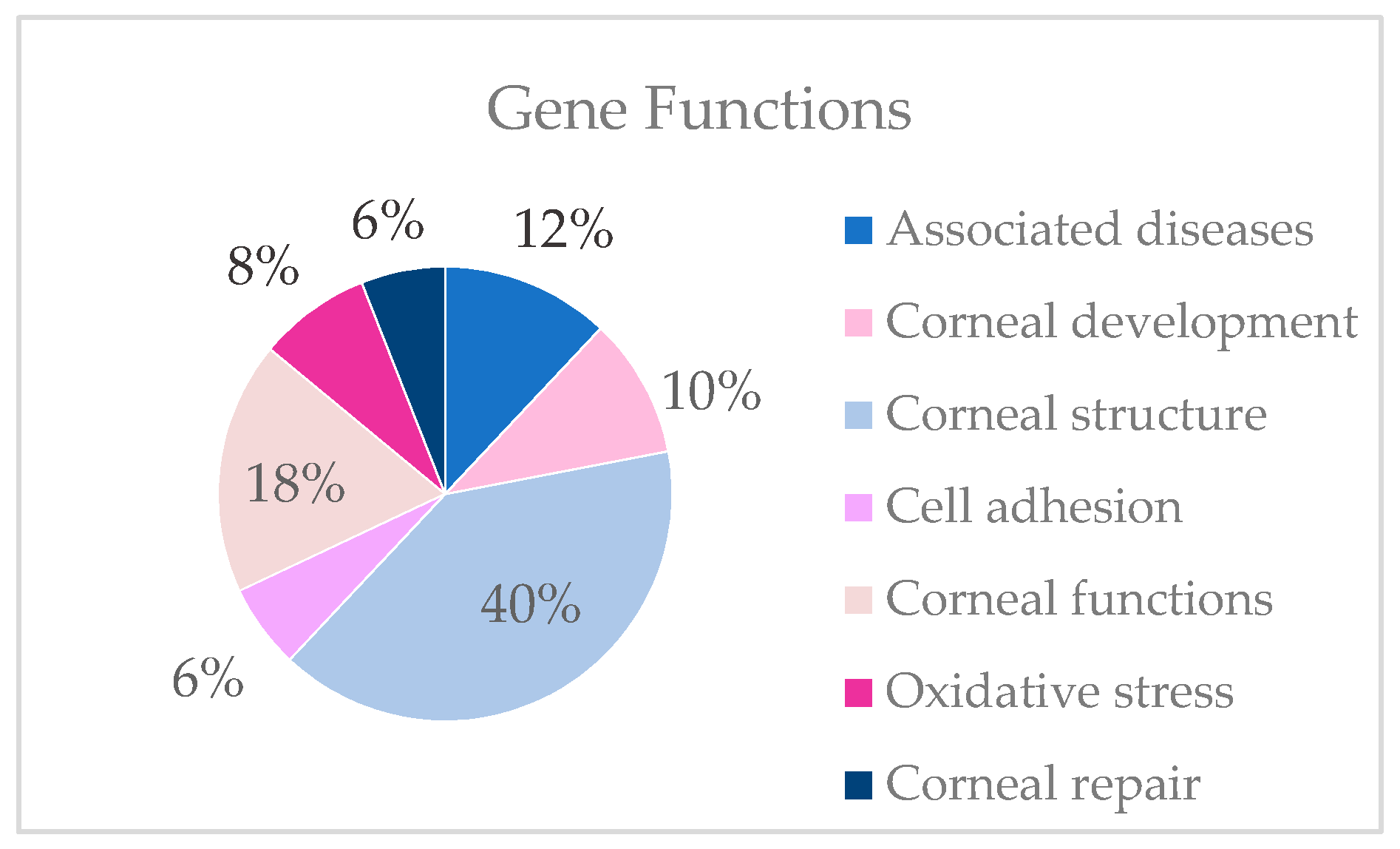
- Genes described in corneal dystrophies and other ocular diseases.
- Genes involved in corneal development.
- Genes involved in corneal structure.
- Genes involved in corneal function.
2. Materials and Methods
2.1. Ophthalmological Studies
2.2. Genetic Studies
3. Results
3.1. Ophthalmological Examination
3.2. Molecular Genetics
4. Discussion
4.1. Ophthalmological Studies
4.2. Genetic Study
- Family OFT-00242.
- Family OFT-00273.
- Family OFT-00290.
- Family OFT-00705.
- Family OFT-00813.
- Family OFT-00814.
- Family OFT-00815.
- Family OFT-00816.
- Family OFT-00817.
- Family OFT-00818.
- Family OFT-00819.
- Family OFT-00820.
- Family OFT-00821.
- Family OFT-00846.
- Family OFT-00847.
- Family OFT-00848.
- Family OFT-00850.
- Family OFT-00851.
- Family OFT-00852.
- Family OFT-00853.
- Family OFT-00854.
- Family OFT-00855.
- Family OFT-00856.
- Family OFT-00857.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family ID | Normal/Topographic/Suspect/Frustre | KC Grade | KC Pattern | Q 4 RE (D) | K1 5 RE (D) | K2 6 RE (D) | Kmed 7 RE (D) | Kmax 8 RE (D) | TP 9 RE (μm) | AT 10 RE (D) | Q LE (D) | K1 LE (D) | K2 LE (D) | Kmed LE (D) | Kmax LE (D) | TP LE (μm) | AT LE (D) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OFT-00242 | Topographic BE 1 | RE 2 1, LE 3 1–2 | Snowman RE, Croissant LE | −0.48 | 42.2 | 44.6 | 43.4 | 47.1 | 529 | 2.4 | −0.61 | 43.8 | 45.6 | 44.7 | 48.1 | 508 | 1.8 |
| OFT-00273 | Topographic BE | RE 2–3, LE 2 | Snowman BE | −1.54 | 41.4 | 52.2 | 46.2 | 57.2 | 501 | 10.8 | −0.49 | 38 | 41.4 | 39.6 | 48 | 500 | 3.4 |
| OFT-00290 | Topographic BE | RE 1–2, LE 3–4 | Croissant RE, Snowman LE | −0.49 | 44 | 46.1 | 45 | 49.5 | 463 | 2.1 | −1.59 | 49.1 | 53.1 | 51 | 67.4 | 433 | 4 |
| OFT-00705 | Topographic BE | RE 1–2, LE 2 | Duck BE | −0.59 | 44.3 | 47.2 | 45.7 | 50 | 523 | 2.9 | −0.84 | 44.6 | 49.7 | 47 | 53.3 | 501 | 5.1 |
| OFT-00813 | Topographic RE, Frustre LE | RE 1, LE 0 | Duck RE | −0.57 | 42.5 | 47.3 | 44.7 | 47.9 | 521 | 4.8 | −0.53 | 42.9 | 46.5 | 44.7 | 47 | 513 | 3.6 |
| OFT-00814 | Topographic BE | RE 3, LE 2 | Duck BE | −0.38 | 42.6 | 47.3 | 44.8 | 56.9 | 452 | 4.7 | −0.04 | 49.4 | 43.3 | 41.8 | 50.8 | 463 | 6.1 |
| OFT-00815 | Topographic BE | RE 3, LE 1 | Nipple RE, Bowtie LE | −1.73 | 52.1 | 55.1 | 53.6 | 67 | 394 | 3 | −0.82 | 45.3 | 47.4 | 46.3 | 49 | 432 | 2.1 |
| OFT-00816 | Topographic BE | RE 2, LE 1–2 | Duck BE | −0.8 | 45.4 | 51.7 | 48.4 | 59.2 | 492 | 6.3 | −0.64 | 45.1 | 49.6 | 47.2 | 53.6 | 505 | 4.5 |
| OFT-00817 | Frustre RE, Normal LE | BE 0 | - | −0.38 | 43.7 | 45.1 | 44.4 | 45.4 | 534 | 1.4 | −0.4 | 43 | 46.2 | 44.5 | 46.5 | 533 | 3.2 |
| OFT-00818 | Topographic BE | RE 3, LE 2 | Duck BE | −1.76 | 45.7 | 50 | 47.8 | 59.8 | 562 | 4.3 | −0.39 | 40.8 | 43.3 | 42 | 48.2 | 575 | 2.5 |
| OFT-00819 | Topographic BE | BE 1 | Croissant RE, Bowtie LE | −0.58 | 45 | 46.8 | 45.9 | 47.2 | 515 | 1.8 | −0.63 | 45.3 | 47.9 | 46.6 | 48.3 | 517 | 2.6 |
| OFT-00820 | Topographic RE, Suspect LE | RE 3 | Duck RE | −0.61 | 49.3 | 57.1 | 52.9 | 79.3 | 432 | 7.8 | −0.39 | 45.1 | 46.1 | 45.6 | 49.1 | 471 | 1 |
| OFT-00821 | Topographic BE | RE 2, LE 1–2 | Snowman BE | −0.51 | 46 | 49 | 47.5 | 55.1 | 532 | 3 | −0.9 | 45.2 | 46.6 | 45.9 | 49.9 | 549 | 1.2 |
| OFT-00846 | Topographic BE | RE 1, LE 2–3 | Snowman RE, Duck LE | −0.88 | 43.4 | 48.5 | 45.8 | 51.3 | 516 | 5.1 | −1.38 | 44.8 | 54.8 | 49.3 | 60.2 | 501 | 10 |
| OFT-00847 | Suspect BE | - | - | −0.5 | 40.1 | 44.3 | 42.1 | 44.9 | 542 | 4.2 | −0.51 | 41.4 | 43.6 | 42.5 | 43.9 | 513 | 2.2 |
| OFT-00848 | Topographic BE | RE 2, LE 1 | Duck BE | −1.38 | 44.9 | 50.7 | 47.6 | 59.1 | 517 | 5.8 | −0.29 | 42.2 | 43.2 | 42.7 | 46 | 556 | 1 |
| OFT-00850 | Topographic BE | RE 3–4, LE 3 | Snowman RE, Croissant LE | −1.79 | 53.2 | 58.2 | 55.6 | 65.9 | 407 | 5 | −1.12 | 47.6 | 50.3 | 48.9 | 57.4 | 437 | 2.7 |
| OFT-00851 | Topographic BE | RE 3, LE 1 | Croissant RE, Duck LE | −0.05 | 42.1 | 46.5 | 44.2 | 54.2 | 478 | 4.4 | −0.21 | 42.9 | 43.8 | 43.4 | 49.5 | 499 | 0.9 |
| OFT-00852 | Topographic BE | RE 2, LE 3–4 | Duck RE, Nipple LE | −0.76 | 44.7 | 48.5 | 46.5 | 54.2 | 477 | 3.8 | −1.3 | 50.6 | 55.3 | 52.9 | 71.5 | 434 | 4.7 |
| OFT-00853 | Topographic BE | RE 2, LE 2–3 | Duck BE | −0.55 | 45.6 | 50.4 | 47.9 | 56.3 | 469 | 4.8 | −0.84 | 47.9 | 52.5 | 50.1 | 61.9 | 464 | 4.6 |
| OFT-00854 | Normal RE, Topographic LE | RE 0, LE 3 | Duck LE | −0.33 | 41.5 | 42.3 | 41.9 | 42.7 | 502 | 0.8 | −0.69 | 42.9 | 48.2 | 45.4 | 59.4 | 443 | 5.3 |
| OFT-00855 | Topographic BE | RE 3, LE 3–4 | Nipple BE | −2.03 | 44.6 | 54.6 | 49.1 | 62.4 | 473 | 10 | −2.65 | 57.2 | 66 | 61.3 | 76.3 | 387 | 8.8 |
| OFT-00856 | Normal RE, Topographic LE | RE 0, LE 1–2 | Duck LE | −0.29 | 45.8 | 46.5 | 46.2 | 47.1 | 502 | 0.7 | −0.75 | 47.1 | 51.5 | 49.2 | 54.2 | 484 | 9.8 |
| OFT-00857 | Topographic BE | RE 2, LE 1 | Snowman BE | −1.01 | 44.2 | 49 | 46.5 | 56.8 | 522 | 4.8 | −0.46 | 41.3 | 43.3 | 42.2 | 46.5 | 544 | 2 |
| Family | Sex | Age | Ethnicity | Family History of KC | Consanguinity | KC Uni/Bilateral | Atopy | Eye Rubbing | Ocular Personal History | Systemic Personal History |
|---|---|---|---|---|---|---|---|---|---|---|
| OFT-00242 | Male | 15 | Spanish | No | No | Bilateral | Yes | Yes | MGD 1 | Overweight |
| OFT-00273 | Male | 13 | Spanish–British | No | No | Bilateral | No | No | No | ADHD 2 |
| OFT-00290 | Male | 14 | Spanish | No | No | Bilateral | No | Yes | Anterior stromal corneal dystrophy | Obesity |
| OFT-00705 | Male | 13 | Spanish | Father | No | Bilateral | Yes | Yes | Atopic blepharitis | No |
| OFT-00813 | Male | 10 | South American | Father | No | Bilateral | Yes | Yes | No | Obesity |
| OFT-00814 | Male | 17 | Spanish | No | No | Bilateral | No | No | No | ADHD |
| OFT-00815 | Male | 14 | Spanish | Mother | No | Bilateral | Yes | Yes | Exophoria RE 3 | Obesity |
| OFT-00816 | Male | 11 | South American | No | No | Bilateral | Yes | Yes | No | Obesity |
| OFT-00817 | Female | 8 | South American | No | No | Unilateral (RE) | Yes | No | Congenital astigmatism LE 4 | Obesity–overweight, precocious puberty |
| OFT-00818 | Male | 12 | Spanish | Father | No | Bilateral | Yes | Yes | MGD | OSA 5, obesity, depression |
| OFT-00819 | Female | 11 | South American | Mother | No | Bilateral | No | No | No | Overweight |
| OFT-00820 | Male | 13 | South American | Mother | No | Bilateral | No | Sí | Retinopathy of prematurity, PPMD 6, MGD | Scoliosis, prematurity |
| OFT-00821 | Male | 15 | Spanish | No | No | Bilateral | Yes | Yes | No | No |
| OFT-00846 | Female | 10 | South American | Mother | No | Bilateral | Yes | Yes | No | Precocious puberty, adjustment disorder |
| OFT-00847 | Female | 8 | Spanish | Father, paternal uncle | No | Bilateral | No | No | No | No |
| OFT-00848 | Male | 11 | Spanish–Portuguese | Father | No | Bilateral | Yes | No | Anisometropia | Obesity–overweight, ADHD |
| OFT-00850 | Male | 13 | Spanish–South American | Mother | No | Bilateral | Yes | No | Anterior blepharitis, MGD | Obesity, anxiety and adjustment disorder |
| OFT-00851 | Female | 16 | Spanish | No | No | Bilateral | Yes | Yes | No | No |
| OFT-00852 | Male | 18 | South American | No | No | Bilateral | No | No | No | No |
| OFT-00853 | Male | 14 | Spanish | Mother, father, maternal aunt and cousin | No | Bilateral | Yes | Yes | No | Overweight |
| OFT-00854 | Male | 17 | Spanish | Sister | No | Unilateral (LE) | Yes | No | Congenital cataracts LE | Pectus Excavatum |
| OFT-00855 | Male | 13 | Spanish | No | No | Bilateral | No | Yes | MGD | Asperger Syndrome, obesity |
| OFT-00856 | Female | 14 | Spanish–South American | No | No | Unilateral (LE) | No | Yes | MGD | Scoliosis, overweight |
| OFT-00857 | Male | 14 | Spanish | No | No | Bilateral | Yes | Yes | No | Obesity |
| Code | Description |
|---|---|
| PVS1 | Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where loss of function is a known mechanism of disease. |
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. |
| PS2 | De novo (both maternity and paternity confirmed) in a patient with the disease and no family history. |
| PS3 | Well-established in vitro or in vivo functional studies supporting a damaging effect on the gene or gene product. |
| BS1 | Allele frequency is greater than expected for the disorder. |
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain (e.g., active site of an enzyme) with no benign variation. |
| PM2 | Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, and Exome Aggregation Consortium. |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant. |
| PM4 | Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants. |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has previously been observed. |
| PP1 | Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease. |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease. |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product (e.g., conservation, evolutionary, splicing impact). |
| PP5 | Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation. |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.) |
| BP6 | Reputable source recently reports variant as benign, but the evidence is not available for the laboratory to perform an independent evaluation. |
References
- Veerappa, A.M. Cascade of interactions between candidate genes reveals convergent mechanisms in keratoconus disease pathogenesis. Ophthalmic Genet. 2021, 42, 114–131. [Google Scholar] [CrossRef]
- Santodomingo-Rubido, J.; Carracedo, G.; Suzaki, A.; Villa-Collar, C.; Vincent, S.J.; Wolffsohn, J.S. Keratoconus: An updated review. Cont. Lens Anterior Eye 2022, 45, 101559. [Google Scholar] [CrossRef] [PubMed]
- Loukovitis, E.; Sfakianakis, K.; Syrmakesi, P.; Tsotridou, E.; Orfanidou, M.; Bakaloudi, D.R.; Stoila, M.; Kozei, A.; Koronis, S.; Zachariadis, Z.; et al. Genetic Aspects of Keratoconus: A Literature Review Exploring Potential Genetic Contributions and Possible Genetic Relationships with Comorbidities. Ophthalmol. Ther. 2018, 7, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Unni, P.; Lee, H.J. Systemic Associations with Keratoconus. Life 2023, 13, 1363. [Google Scholar] [CrossRef]
- Asimellis, G.; Kaufman, E.J. Keratoconus; StatPearls: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470435/ (accessed on 28 August 2023).
- Ferrari, G.; Rama, P. The keratoconus enigma: A review with emphasis on pathogenesis. Ocul. Surf. 2020, 18, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Bataille, L. Guía de Actuación en el Queratocono. 2015. Available online: http://www.oftared.com/docs/0b19db.pdf (accessed on 20 May 2023).
- Volatier, T.L.A.; Figueiredo, F.C.; Connon, C.J. Keratoconus at a Molecular Level: A Review. Anat. Rec. 2020, 303, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Keratoconus—About the Disease—Genetic and Rare Diseases Information Center. Available online: https://rarediseases.info.nih.gov/diseases/6824/keratoconus (accessed on 28 August 2023).
- Fernández Berdasco, K.; Alfaya Muñoz, L.B.; Corzo Fernández, C.R.; Señaris González, A.; Baamonde Arbaiza, B. Clinical-epidemiological characteristics of keratoconus in Asturias. Arch. Soc. Esp. Oftalmol. 2023, 98, 65–71. [Google Scholar] [CrossRef]
- Buzzonetti, L.; Bohringer, D.; Liskova, P.; Lang, S.; Valente, P. Keratoconus in Children: A Literature Review. Cornea 2020, 39, 1592–1598. [Google Scholar] [CrossRef]
- Anitha, V.; Vanathi, M.; Raghavan, A.; Rajaraman, R.; Ravindran, M.; Tandon, R. Pediatric keratoconus—Current perspectives and clinical challenges. Indian J. Ophthalmol. 2021, 69, 214–225. [Google Scholar] [CrossRef]
- Rocha de Lossada, C. Caracterización del Queratocono Infantil y Juvenil en un Estudio Multicéntrico. 2021. Available online: https://riuma.uma.es/xmlui/handle/10630/24194 (accessed on 3 June 2023).
- Fernández-Vega Cueto, L. Clasificación del Queratocono Para su Corrección Quirúrgica con Segmentos de Anillo Intracorneales tipo Ferrara. 2016. Available online: https://dialnet.unirioja.es/servlet/tesis?codigo=83581&info=resumen&idioma=SPA (accessed on 3 June 2023).
- Gupta, Y.; Saxena, R.; Jhanji, V.; Maharana, P.K.; Sinha, R.; Agarwal, T.; Titiyal, J.S.; Sharma, N. Management Outcomes in Pediatric Keratoconus: Childhood Keratoconus Study. J. Ophthalmol. 2022, 2022, 4021288. [Google Scholar] [CrossRef]
- Eghrari, A.O.; Riazuddin, S.A.; Gottsch, J.D. Overview of the Cornea: Structure, Function, and Development. Prog. Mol. Biol. Transl. Sci. 2015, 134, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.E.M.; Burdon, K.P. Genetic and Environmental Risk Factors for Keratoconus. Annu. Rev. Vis. Sci. 2020, 6, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Loh, I.P.; Sherwin, T. Is Keratoconus an Inflammatory Disease? The Implication of Inflammatory Pathways. Ocul. Immunol. Inflamm. 2020, 30, 246–255. [Google Scholar] [CrossRef]
- di Martino, E.; Ali, M.; Inglehearn, C.F. Matrix metalloproteinases in keratoconus—Too much of a good thing? Exp. Eye Res. 2019, 182, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Bykhovskaya, Y.; Li, X.; Epifantseva, I.; Haritunians, T.; Siscovick, D.; Aldave, A.; Szczotka-Flynn, L.; Iyengar, S.K.; Taylor, K.D.; Rotter, J.I.; et al. Variation in the lysyl oxidase (LOX) gene is associated with keratoconus in family-based and case-control studies. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4152–4157. [Google Scholar] [CrossRef]
- Hashemi, H.; Heydarian, S.; Hooshmand, E.; Saatchi, M.; Yekta, A.; Aghamirsalim, M.; Valadkhan, M.; Mortazavi, M.; Hashemi, A.; Khabazkhoob, M. The Prevalence and Risk Factors for Keratoconus: A Systematic Review and Meta-Analysis. Cornea 2020, 39, 263. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yuan, Y.; Sun, T.; Zhang, Y.; Chen, Y. Associations Between Keratoconus and the Level of Sex Hormones: A Cross-Sectional Study. Front. Med. 2022, 9, 828233. [Google Scholar] [CrossRef]
- Chen, B.; Yu, X.; Zhang, X.; Yang, H.; Cui, Y.; Shentu, X. Novel Mutations Identified in the Chinese Han Population with Keratoconus by Next-Generation Sequencing. J. Ophthalmol. 2022, 2022, 9991910. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, B.; Ma, L.; Qin, Y.; Li, S.; Sun, C.; Liang, J.; Han, Y.; Zhuang, W. Clinical and genetic analysis VSX1 variants among families with keratoconus in northwest China. Front. Genet. 2023, 14, 1145426. [Google Scholar] [CrossRef]
- Stabuc-Silih, M.; Ravnik-Glavac, M.; Glavac, D.; Hawlina, M.; Strazisar, M. Polymorphisms in COL4A3 and COL4A4 genes associated with keratoconus. Mol. Vis. 2009, 15, 2848–2860. Available online: https://pubmed.ncbi.nlm.nih.gov/20029656/ (accessed on 29 August 2023).
- Li, X.; Bykhovskaya, Y.; Canedo, A.L.C.; Haritunians, T.; Siscovick, D.; Aldave, A.J.; Szczotka-Flynn, L.; Iyengar, S.K.; Rotter, J.I.; Taylor, K.D.; et al. Genetic association of COL5A1 variants in keratoconus patients suggests a complex connection between corneal thinning and keratoconus. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Karolak, J.A.; Rydzanicz, M.; Ginter-Matuszewska, B.; Pitarque, J.A.; Molinari, A.; Bejjani, B.A.; Gajecka, M. Variant c.2262A>C in DOCK9 Leads to Exon Skipping in Keratoconus Family. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7687–7690. [Google Scholar] [CrossRef] [PubMed]
- Atilano, S.; Lee, D.; Fukuhara, P.; Chwa, M.; Nesburn, A.; Udar, N.; Kenney, C. Corneal Oxidative Damage in Keratoconus Cells due to Decreased Oxidant Elimination from Modified Expression Levels of SOD Enzymes, PRDX6, SCARA3, CPSF3, and FOXM1. J. Ophthalmic Vis. Res. 2019, 14, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Franklin. Available online: https://franklin.genoox.com/clinical-db/home (accessed on 29 August 2023).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- PubMed. 2023. Available online: https://pubmed.ncbi.nlm.nih.gov/ (accessed on 29 August 2023).
- GeneCards—Human Genes|Gene Database|Gene Search. 2023. Available online: https://www.genecards.org/ (accessed on 3 June 2023).
- Home—OMIM. Available online: https://www.omim.org/ (accessed on 29 August 2023).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 29 August 2023).
- gnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 29 August 2023).
- UniProt. Available online: https://www.uniprot.org/ (accessed on 29 August 2023).
- GeneMatcher (GM). Available online: https://genematcher.org/ (accessed on 29 August 2023).
- Fransen, E.; Valgaeren, H.; Janssens, K.; Sommen, M.; De Ridder, R.; Vandeweyer, G.; Bisceglia, L.; Soler, V.; Hoischen, A.; Mortier, G.; et al. Resequencing of candidate genes for Keratoconus reveals a role for Ehlers–Danlos Syndrome genes. Eur. J. Hum. Genet. 2021, 29, 1745–1755. [Google Scholar] [CrossRef]
- Yu, X.; Chen, B.; Zhang, X.; Shentu, X. Identification of seven novel ZNF469 mutations in keratoconus patients in a Han Chinese population. Mol. Vis. 2017, 23, 296. [Google Scholar]
- Xu, L.; Yang, K.; Zhu, M.; Yin, S.; Gu, Y.; Fan, Q.; Wang, Y.; Pang, C.; Ren, S. Trio-based exome sequencing broaden the genetic spectrum in keratoconus. Exp. Eye Res. 2023, 226, 109342. [Google Scholar] [CrossRef]
- Kuot, A.; Mills, R.; Craig, J.E.; Sharma, S.; Burdon, K.P. Screening of the COL8A2 gene in an Australian family with early-onset Fuchs’ endothelial corneal dystrophy. Clin. Exp. Ophthalmol. 2014, 42, 198–200. [Google Scholar] [CrossRef]
- Hao, X.D.; Chen, X.N.; Zhang, Y.Y.; Chen, P.; Wei, C.; Shi, W.Y.; Gao, H. Multi-level consistent changes of the ECM pathway identified in a typical keratoconus twin’s family by multi-omics analysis. Orphanet J. Rare Dis. 2020, 15, 227. [Google Scholar] [CrossRef]
- Purcell, J.J.; Krachmer, J.H.; Weingeist, T.A. Fleck corneal dystrophy. Arch. Ophthalmol. 1977, 95, 440–444. [Google Scholar] [CrossRef]
- Cheng, W.Y.; Yang, S.Y.; Huang, X.Y.; Zi, F.Y.; Li, H.P.; Sheng, X.L. Identification of genetic variants in five chinese families with keratoconus: Pathogenicity analysis and characteristics of parental corneal topography. Front. Genet. 2022, 13, 978684. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, Y.S.; Galvis, V.; Tello, A.; Rueda, D.; García, J.D. Genetics vs chronic corneal mechanical trauma in the etiology of keratoconus. Exp. Eye Res. 2021, 202, 108328. [Google Scholar] [CrossRef]
- Khaled, M.L.; Bykhovskaya, Y.; Gu, C.; Liu, A.; Drewry, M.D.; Chen, Z.; Mysona, B.A.; Parker, E.; McNabb, R.P.; Yu, H.; et al. PPIP5K2 and PCSK1 are Candidate Genetic Contributors to Familial Keratoconus. Sci. Rep. 2019, 9, 19406. [Google Scholar] [CrossRef] [PubMed]
- Arce-González, R.; Chacon-Camacho, O.F.; Ordoñez-Labastida, V.; Graue-Hernandez, E.O.; Navas-Pérez, A.; Zenteno, J.C. A novel homozygous ZNF469 variant causing brittle cornea syndrome is associated with corneal ectasias in heterozygous carriers. Int. Ophthalmol. 2023, 43, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, E.; Bardak, H.; Gunay, M.; Bardak, Y.; Imamoglu, S.; Ozbas, H.; Bagci, O. Novel Zinc Finger Protein Gene 469 (ZNF469) Variants in Advanced Keratoconus. Curr. Eye Res. 2017, 42, 1396–1400. [Google Scholar] [CrossRef]
- Shinde, V.; Sobreira, N.; Wohler, E.S.; Maiti, G.; Hu, N.; Silvestri, G.; George, S.; Jackson, J.; Chakravarti, A.; Willoughby, C.E.; et al. Pathogenic alleles in microtubule, secretory granule and extracellular matrix-related genes in familial keratoconus. Hum. Mol. Genet. 2021, 30, 658. [Google Scholar] [CrossRef]
- Xu, L.; Yang, K.; Yin, S.; Gu, Y.; Fan, Q.; Wang, Y.; Zhao, D.; Ren, S. Family-based exome sequencing identifies candidate genes related to keratoconus in Chinese families. Front. Genet. 2022, 13, 988620. [Google Scholar] [CrossRef]
- Ren, S.; Yang, K.; Fan, Q.; Wang, Q.; Zhu, M.; Yin, S.; Gu, Y.; Xu, L. Bioinformatics analysis of key candidate genes and pathways in Chinese patients with keratoconus. Exp. Eye Res. 2023, 231, 109488. [Google Scholar] [CrossRef]
- McKay, T.B.; Priyadarsini, S.; Rowsey, T.; Karamichos, D. Arginine supplementation promotes extracellular matrix and metabolic changes in keratoconus. Cells 2021, 10, 2076. [Google Scholar] [CrossRef]
- Fernández-Gutiérrez, E.; Fernández-Pérez, P.; Boto-De-Los-Bueis, A.; García-Fernández, L.; Rodríguez-Solana, P.; Solís, M.; Vallespín, E. Posterior Polymorphous Corneal Dystrophy in a Patient with a Novel ZEB1 Gene Mutation. Int. J. Mol. Sci. 2022, 24, 209. [Google Scholar] [CrossRef]
- Amit, C.; Padmanabhan, P.; Narayanan, J. Deciphering the mechanoresponsive role of β-catenin in keratoconus epithelium. Sci. Rep. 2020, 10, 21382. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.; Dash, D.P.; Muszynska, D.; Hosseini, M.; Segev, F.; George, S.; Frazer, D.G.; Moore, J.E.; Kaye, S.B.; Young, T.; et al. Mutational spectrum of the ZEB1 gene in corneal dystrophies supports a genotype-phenotype correlation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.E.; Bradley, D.T.; Campbell, M.; Lechner, J.; Dash, D.P.; Simpson, D.A.; Willoughby, C.E. Mutation altering the miR-184 seed region causes familial keratoconus with cataract. Am. J. Hum. Genet. 2011, 89, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Bower, J.J.; Llanga, T.; Salmon, J.H.; Hirsch, M.L.; Gilger, B.C. Ocular Tolerability and Immune Response to Corneal Intrastromal AAV- IDUA Gene Therapy in New Zealand White Rabbits. Mol. Ther. Methods Clin. Dev. 2020, 18, 24–32. [Google Scholar] [CrossRef]
- Mok, J.W.; So, H.R.; Ha, M.J.; Na, K.S.; Joo, C.K. Association with Corneal Remodeling Related Genes, ALDH3A1, LOX, and SPARC Genes Variations in Korean Keratoconus Patients. Korean J. Ophthalmol. 2021, 35, 120. [Google Scholar] [CrossRef]
- Wojcik, K.A.; Synowiec, E.; Kaminska, A.; Izdebska, J.; Polakowski, P.; Pawlowska, E.; Blasiak, J.; Szaflik, J.; Szaflik, J.P. Polymorphism of the APEX nuclease 1 gene in keratoconus and Fuchs endothelial corneal dystrophy. Cell. Mol. Biol. Lett. 2015, 20, 48–65. [Google Scholar] [CrossRef]
- Akoto, T.; Li, J.J.; Estes, A.J.; Karamichos, D.; Liu, Y. The Underlying Relationship between Keratoconus and Down Syndrome. Int. J. Mol. Sci. 2022, 23, 10796. [Google Scholar] [CrossRef]
- Xu, L.; Yang, K.; Fan, Q.; Gu, Y.; Zhang, B.; Pang, C.; Ren, S. Exome sequencing identification of susceptibility genes in Chinese patients with keratoconus. Ophthalmic Genet. 2020, 41, 518–525. [Google Scholar] [CrossRef]
- Kumar, S.; Dollé, P.; Ghyselinck, N.B.; Duester, G. Endogenous retinoic acid signaling is required for maintenance and regeneration of cornea. Exp. Eye Res. 2017, 154, 190–195. [Google Scholar] [CrossRef]
- Froukh, T.; Hawwari, A. Autosomal Recessive Non-syndromic Keratoconus: Homozygous Frameshift Variant in the Candidate Novel Gene GALNT14. Curr. Mol. Med. 2019, 19, 683–687. [Google Scholar] [CrossRef]
- Khawaja, A.P.; Rojas Lopez, K.E.; Hardcastle, A.J.; Hammond, C.J.; Liskova, P.; Davidson, A.E.; Gore, D.M.; Hafford Tear, N.J.; Pontikos, N.; Hayat, S.; et al. Genetic Variants Associated with Corneal Biomechanical Properties and Potentially Conferring Susceptibility to Keratoconus in a Genome-Wide Association Study. JAMA Ophthalmol. 2019, 137, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Cehajic-Kapetanovic, J.; Xue, K.; Martinez-Fernandez de la Camara, C.; Nanda, A.; Davies, A.; Wood, L.J.; Paola Salvetti, A.; Fischer, M.D.; Aylward, J.W.; Barnard, A.R.; et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 2020, 26, 354–359. [Google Scholar] [CrossRef] [PubMed]




| Grade | Visual Acuity with Glasses | Visual Acuity with CL 1 | Corneal Indices | Ecc. 30 4 | RMin 5 | Cornea | |
|---|---|---|---|---|---|---|---|
| ISV 2 | KJ 3 | ||||||
| Suspect | 1.0 1.0–1.25 20/15 | 1.0 1.0–1.25 20/15 | <30 | 1.04 1.04–1.07 1.07 | 4 normal values | 7.8 7.8–6.7 6.7 | Clear, inconspicuous cornea. Horizontal, oval or with circular shadows centered or slightly off-center. |
| Grade 1 | 20/25 0.8–1.25 20/15 | 1.0 | 30 30/55 55 | 1.07 1.07–1.15 1.15 | At least 1 value is rarely abnormal | 7.5 7.5–6.5 6.5 | Transparent cornea. Fleischer’s ring at the base of the apex. Decrease in the apex vertex. |
| Grade 2 | 20/60 0.32–1.0 1.0 | 20/30 0.63–1.0 1.0 | 55 55–90 90 | 1.10 1.10–1.25 1.25 | At least 1 value is frequently abnormal | 6.9 6.9–5.3 5.3 | Cornea often remains transparent. The apex occasionally becomes somewhat decentered. Partial or circular Fleischer rings. Occasionally, Vogt’s striae are present. |
| Grade 3 | 20/125 0.16–0.63 20/30 | 20/40 0.5–1.0 1.0 | 90 90–150 150 | 1.15 1.15–1.45 1.45 | 1 value is always abnormal | 6.6 6.6–4.8 4.8 | The apex has decreased in thickness, is off-center and is slightly dark. Fleischer rings clearly visible and circular. Easily identifiable Vogt’s striae. Eventually signs of Munson. |
| Grade 4 | <20/400 <0.05–0.2 20/100 | 20/100 0.2–0.5 20/40 | >150 | >1.50 | 1 value is always abnormal | <5 | The cornea is often scarred. Munson’s sign is evident. |
| Family ID | Keratoconus | Sex | Age | Family History | Genetic Study | |||
|---|---|---|---|---|---|---|---|---|
| RE 1 | LE 2 | |||||||
| Grade | Morphology | Grade | Morphology | |||||
| OFT-00242 | 1 | Snowman | 1–2 | Croissant | M 3 | 15 | No | Proband |
| OFT-00273 | 2–3 | Snowman | 2 | Snowman | M | 13 | No | Proband |
| OFT-00290 | 1–2 | Duck | 3–4 | Snowman | M | 14 | Mother with FCD 5. | Proband + mother |
| OFT-00705 | 1–2 | Duck | 2 | Duck | M | 13 | Father with KC 6 BE 7. | Proband + parents |
| OFT-00813 | 1 | Duck | Frustre | X | M | 10 | Father with suspect KC RE. | Proband + parents |
| OFT-00814 | 3 | Duck | 1 | Duck | M | 17 | No | Proband + parents |
| OFT-00815 | 3 | Nipple | 1 | Bowtie | M | 14 | Mother with KC BE. Grandmother and maternal uncles with astigmatism. | Proband + parents |
| OFT-00816 | 2 | Duck | 1–2 | Duck | M | 11 | No | Proband + parents |
| OFT-00817 | Frustre | - | - | - | F 4 | 8 | No | Proband + parents |
| OFT-00818 | 3 | Duck | 2 | Duck | M | 12 | Father with suspect KC RE and topographic LE. | Proband + father |
| OFT-00819 | 1 | Croissant | 1 | Bowtie | F | 11 | Mother with suspect KC BE. | Proband + mother |
| OFT-00820 | 3 | Duck | Suspect | Snowman | M | 13 | Mother with suspect KC BE. | Proband + parents |
| OFT-00821 | 2 | Snowman | 1–2 | Snowman | M | 15 | No | Proband + parents |
| OFT-00846 | 1 | Snowman | 2–3 | Duck | F | 10 | Mother with frustre KC BE. | Proband + parents |
| OFT-00847 | Suspect | Croissant | Suspect | Croissant | F | 8 | Father with suspect KC LE. Paternal uncle with topographic KC. | Proband + parents |
| OFT-00848 | 3 | Duck | 1 | Duck | M | 11 | Father with frustre KC BE. | Proband + parents |
| OFT-00850 | 3–4 | Snowman | 3 | Duck- Croissant | M | 13 | Mother with suspect KC BE. | Proband + parents |
| OFT-00851 | 3 | Croissant | 1 | Duck | F | 16 | No | Proband + parents |
| OFT-00852 | 2 | Duck | 3–4 | Nipple | M | 18 | No | Proband |
| OFT-00853 | 2 | Duck | 2–3 | Duck | M | 14 | Father with suspect KC BE, mother with suspect KC LE. Maternal cousin with topographic KC BE. | Proband + parents |
| OFT-00854 | - | - | 3 | Duck | M | 17 | Sister with suspect KC. | Proband + parents |
| OFT-00855 | 3 | Nipple | 3–4 | Nipple | M | 13 | No | Proband |
| OFT-00856 | - | - | 1–2 | Duck | F | 14 | No | Proband + parents |
| OFT-00857 | 2 | Snowman | 1 | Snowman | M | 14 | Maternal uncle with KC. | Proband + mother |
| Family | Gene | Mutation | ACMG * Result | Zygosity | Inheritance | De Novo/Inherited |
|---|---|---|---|---|---|---|
| OFT-00242 | ZNF469 | NM_001127464.2:c.11173G>A p.(Ala3725Thr) | VUS 1 | Het 3 | AR 4 | Unknown |
| NDRG1 | NM_001135242.1:c.699-2A>G | LP 2 | Het | AR | Unknown | |
| LOXHD1 | NM_144612.6:c.1843C>T p.(Arg615Trp) | VUS | Het | AR | Unknown | |
| OFT-00273 | MMP1 | NM_002421.3:c.1301G>T p.(Gly434Val) | VUS | Het | Unknown | Unknown |
| MMP2 | NM_004530.5:c.586G>A p.(Ala196Thr) | VUS | Het | AR | Unknown | |
| COL8A2 | NM_005202.3:c.567A>T p.(Glu189Asp) | VUS | Het | AR | Unknown | |
| COL4A3 | NM_000091.4:c.4523A>G p.(Asn1508Ser) | VUS | Het | AR | Unknown | |
| OFT-00290 | PIKFYVE | NM_015040.3:c.3791+2T>C | LP | Het | AD 5 | Maternal |
| LOXL2 | NM_002318.2:c.1642C>A p.(Pro548Thr) | VUS | Het | Unknown | Maternal | |
| OFT-00705 | PPIP5K2 | NM_015216.4:c.86A>C p.(Asp29Ala) | VUS | Het | Unknown | Paternal |
| SLC8A3 | NM_182932.2:c.56T>C p.(Val19Ala) | VUS | Het | Unknown | Paternal | |
| OFT-00813 | ZNF469 | NM_001127464.2:c.9347C>T p.(Thr3116Ile) | VUS | Het | AR | Paternal |
| TNXB | NM_019105.6:c.4010G>A p.(Arg1337His) | VUS | Het | AD/AR | Paternal | |
| HSPG2 | NM_005529.6:c.2023C>T p.(Arg675Trp) | VUS | Het | AD/AR | Maternal | |
| OFT-00814 | LOXHD1 | NM_144612.6:c.740C>A p.(Ala247Glu) | VUS | Het | AR | Paternal |
| TTN | NM_133378.4:c.91524G>C p.(Glu30508Asp) | VUS | Het | AD | Maternal | |
| OFT-00815 | COL6A3 | NM_004369.3:c.8865dupC p.(Ala2956fs) | LP | Het | AD/AR | Paternal |
| COL6A3 | NM_004369.3:c.5681C>T p.(Pro1894Leu) | VUS | Het | AD/AR | Maternal | |
| COL4A4 | NM_000092.4:c.4291C>T p.(Arg1431Cys) | VUS | Het | AD/AR | Maternal | |
| COL2A1 | NM_033150.2:c.402G>A p.(Met134Ile) | VUS | Het | AD | Maternal | |
| TTN | NM_133378.4:c.64442T>C p.(Leu21481Pro) | VUS | Het | AD | Maternal | |
| OFT-00816 | HSPG2 | NM_005529.6:c.10084C>T p.(Arg3362Cys) | VUS | Het | AD/AR | Maternal |
| COL4A2 | NM_001846.3:c.3635-3C>T | VUS | Het | AD | Maternal | |
| COL23A1 | NM_173465.3:c.526G>T p.(Asp176Tyr) | VUS | Het | Unknown | No Maternal | |
| ARG1 | NM_000045.3:c.386T>C p.(Ile129Thr) | VUS | Het | AR | Maternal | |
| DUXA | NM_001012729.1:c.118C>T p.(Pro40Ser) | VUS | Het | Unknown | No Maternal | |
| OFT-00817 | EML6 | NM_001039753.2:c.2347G>T p.(Asp783Tyr) | VUS | Het | Unknown | Maternal |
| SLC4A4 | NM_001098484.2:c.1256C>T p.(Thr419Met) | VUS | Het | AD/AR | Maternal | |
| CPSF3 | NM_016207.3:c.1120A>G p.(Ile374Val) | VUS | Het | Unknown | Paternal | |
| CDH23 | NM_022124.5:c.7834G>T p.(Val2612Leu) | VUS | Het | AD/AR | Maternal | |
| CDHR1 | NM_033100.3:c.1133G>A p.(Arg378Gln) | VUS | Het | AR | Paternal | |
| OFT-00818 | ZEB1 | NM_030751.5:c.2184A>C p.(Glu728Asp) | VUS | Het | AD | Paternal |
| ITGB4 | NM_001005619.1:c.916T>C p.(Ser306Pro) | VUS | Het | AD/AR | Paternal | |
| INPPL1 | NM_001567.3:c.2659+4G>C | VUS | Het | AR | No Paternal | |
| IDUA | NM_000203.4:c.299+2092C>T | VUS | Het | AR | Paternal | |
| OFT-00819 | INPPL1 | NM_001567.3:c.3584G>T p.(Gly1195Val) | VUS | Het | AR | Maternal |
| GUCY2D | NM_000180.3:c.1720C>T p.(Arg574Cys) | VUS | Het | AD/AR | Maternal | |
| CDH23 | NM_022124.5:c.7351A>G p.(Asn2451Asp) | VUS | Het | AD/AR | Maternal | |
| CDHR1 | NM_033100.3:c.931C>A p.(Leu311Ile) | VUS | Het | AR | Maternal | |
| OFT-00820 | HSPG2 | NM_005529.6:c.9476G>A p.(Arg3159Gln) | VUS | Het | AD/AR | Maternal |
| IMPG2 | NM_016247.3:c.3688G>T p.(Ala1230Ser) | VUS | Het | AR | Maternal | |
| CPAMD8 | NM_015692.2:c.4914+2_4914+3insT | LP | Het | AR | Paternal | |
| OFT-00821 | INPPL1 | NM_001567.3:c.2251G>C p.(Glu751Gln) | VUS | Het | AR | Maternal |
| IDUA | NM_000203.4:c.806C>G p.(Ser269Cys) | VUS | Het | AR | Paternal | |
| OFT-00846 | ITGB4 | NM_001005619.1:c.3265G>C p.(Gly1089Arg) | VUS | Het | AD/AR | Maternal |
| SPARC | NM_003118.3:c.280G>A p.(Val94Met) | VUS | Het | AD/AR | Maternal | |
| TTN | NM_133378.4:c.73585G>C p.(Val24529Leu) | VUS | Het | AD | No Maternal | |
| OFT-00847 | KRT7 | NM_005556.3:c.73C>A p.(Arg25Ser) | VUS | Het | Unknown | Paternal |
| KRT19 | NM_002276.4:c.913G>A p.(Gly305Ser) | VUS | Het | Unknown | Paternal | |
| OFT-00848 | IDUA | NM_000203.4:c.30_31insCTGGCGCTC p.(Leu11_Ala12insAlaLeuLeu) | VUS | Het | AR | Paternal |
| APEX1 | NM_001641.3:c.662G>A p.(Arg221His) | VUS | Het | Unknown | Paternal | |
| OFT-00850 | COL6A5 | NM_001278298.1:c.5392G>A p.(Gly1798Arg) | VUS | Het | Unknown | Maternal |
| GRHL1 | NM_198182.2:c.860T>G p.(Val287Gly) | VUS | Het | Unknown | Maternal | |
| SLC4A4 | NM_001098484.2:c.149G>C p.(Gly50Ala) | VUS | Het | AD/AR | Maternal | |
| OFT-00851 | EML6 | NM_001039753.2:c.3181G>A p.(Asp1061Asn) | VUS | Het | Unknown | Maternal |
| COL9A3 | NM_001853.3:c.736C>T p.(Arg246Trp) | VUS | Het | AD | Paternal | |
| TTN | NM_133378.4:c.10361-3546T>C | VUS | Het | AR | Paternal | |
| OFT-00852 | SLC4A11 | NM_001174090.1:c.2305G>A p.(Gly769Arg) | VUS | Het | AD | Unknown |
| LRP1B | NM_018557.2:c.12983A>G p.(Tyr4328Cys) | VUS | Het | Unknown | Unknown | |
| PIK3CG | NM_002649.3:c.850G>A p.(Glu284Lys) | VUS | Het | Unknown | Unknown | |
| COL6A6 | NM_001102608.1:c.5497T>C p.(Tyr1833His) | VUS | Het | Unknown | Unknown | |
| LOXHD1 | NM_144612.6:c.2353G>A p.(Ala785Thr) | VUS | Het | AR | Unknown | |
| OFT-00853 | LRP1B | NM_018557.2:c.2568C>G p.(His856Gln) | VUS | Het | Unknown | Paternal |
| ALDH1A2 | NM_003888.3:c.173A>G p.(Tyr58Cys) | VUS | Het | Unknown | Maternal | |
| CDHR1 | NM_033100.3:c.1397A>G p.(Asn466Ser) | VUS | Het | AR | Paternal | |
| OFT-00854 | IPO5 | NM_002271.4:c.929C>T p.(Ala310Val) | VUS | Het | Unknown | Maternal |
| GALNT14 | NM_024572.3:c.472G>A p.(Asp158Asn) | VUS | Het | Unknown | Maternal | |
| COL8A2 | NM_005202.3:c.1645G>A p.(Gly549Ser) | VUS | Het | AD | Maternal | |
| OFT-00855 | ADAMTS9 | NM_182920.1:c.3967C>T p.(Arg1323Cys) | VUS | Het | Unknown | Unknown |
| ADAMTS8 | NM_007037.5:c.707G>A p.(Gly236Glu) | VUS | Het | Unknown | Unknown | |
| OFT-00856 | EML6 | NM_001039753.2:c.2752A>G p.(Thr918Ala) | VUS | Het | Unknown | Paternal |
| DOCK9 | NM_001130048.1:c.5018G>A p.(Arg1673Gln) | VUS | Het | Unknown | Maternal | |
| ADAMTS7 | NM_014272.4:c.646C>T p.(Arg216Trp) | VUS | Het | Unknown | Paternal | |
| COL5A3 | NM_015719.3:c.2194G>A p.(Glu732Lys) | VUS | Het | Unknown | Paternal | |
| OFT-00857 | ZNF469 | NM_001127464.2:c.1475G>A p.(Arg492Gln) | VUS | Het | AR | No Maternal |
| IDUA | NM_000203.4:c.1354C>A p.(Arg452Ser) | VUS | Het | AR | Maternal | |
| LOXL1 | NM_005576.3:c.1672C>G p.(His558Asp) | VUS | Het | Unknown | Maternal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Atienza, C.; Sánchez-Cazorla, E.; Villoldo-Fernández, N.; del Hierro, A.; Boto, A.; Guerrero-Carretero, M.; Nieves-Moreno, M.; Arruti, N.; Rodríguez-Solana, P.; Mena, R.; et al. Whole-Exome Sequencing of 24 Spanish Families: Candidate Genes for Non-Syndromic Pediatric Keratoconus. Genes 2023, 14, 1838. https://doi.org/10.3390/genes14101838
González-Atienza C, Sánchez-Cazorla E, Villoldo-Fernández N, del Hierro A, Boto A, Guerrero-Carretero M, Nieves-Moreno M, Arruti N, Rodríguez-Solana P, Mena R, et al. Whole-Exome Sequencing of 24 Spanish Families: Candidate Genes for Non-Syndromic Pediatric Keratoconus. Genes. 2023; 14(10):1838. https://doi.org/10.3390/genes14101838
Chicago/Turabian StyleGonzález-Atienza, Carmen, Eloísa Sánchez-Cazorla, Natalia Villoldo-Fernández, Almudena del Hierro, Ana Boto, Marta Guerrero-Carretero, María Nieves-Moreno, Natalia Arruti, Patricia Rodríguez-Solana, Rocío Mena, and et al. 2023. "Whole-Exome Sequencing of 24 Spanish Families: Candidate Genes for Non-Syndromic Pediatric Keratoconus" Genes 14, no. 10: 1838. https://doi.org/10.3390/genes14101838
APA StyleGonzález-Atienza, C., Sánchez-Cazorla, E., Villoldo-Fernández, N., del Hierro, A., Boto, A., Guerrero-Carretero, M., Nieves-Moreno, M., Arruti, N., Rodríguez-Solana, P., Mena, R., Rodríguez-Jiménez, C., Rosa-Pérez, I., Acal, J. C., Blasco, J., Naranjo-Castresana, M., Ruz-Caracuel, B., Montaño, V. E. F., Ortega Patrón, C., Rubio-Martín, M. E., ... Vallespín, E. (2023). Whole-Exome Sequencing of 24 Spanish Families: Candidate Genes for Non-Syndromic Pediatric Keratoconus. Genes, 14(10), 1838. https://doi.org/10.3390/genes14101838