
Understanding Arrhythmogenic Cardiomyopathy: Advances through the Use of Human Pluripotent Stem Cell Models
Abstract
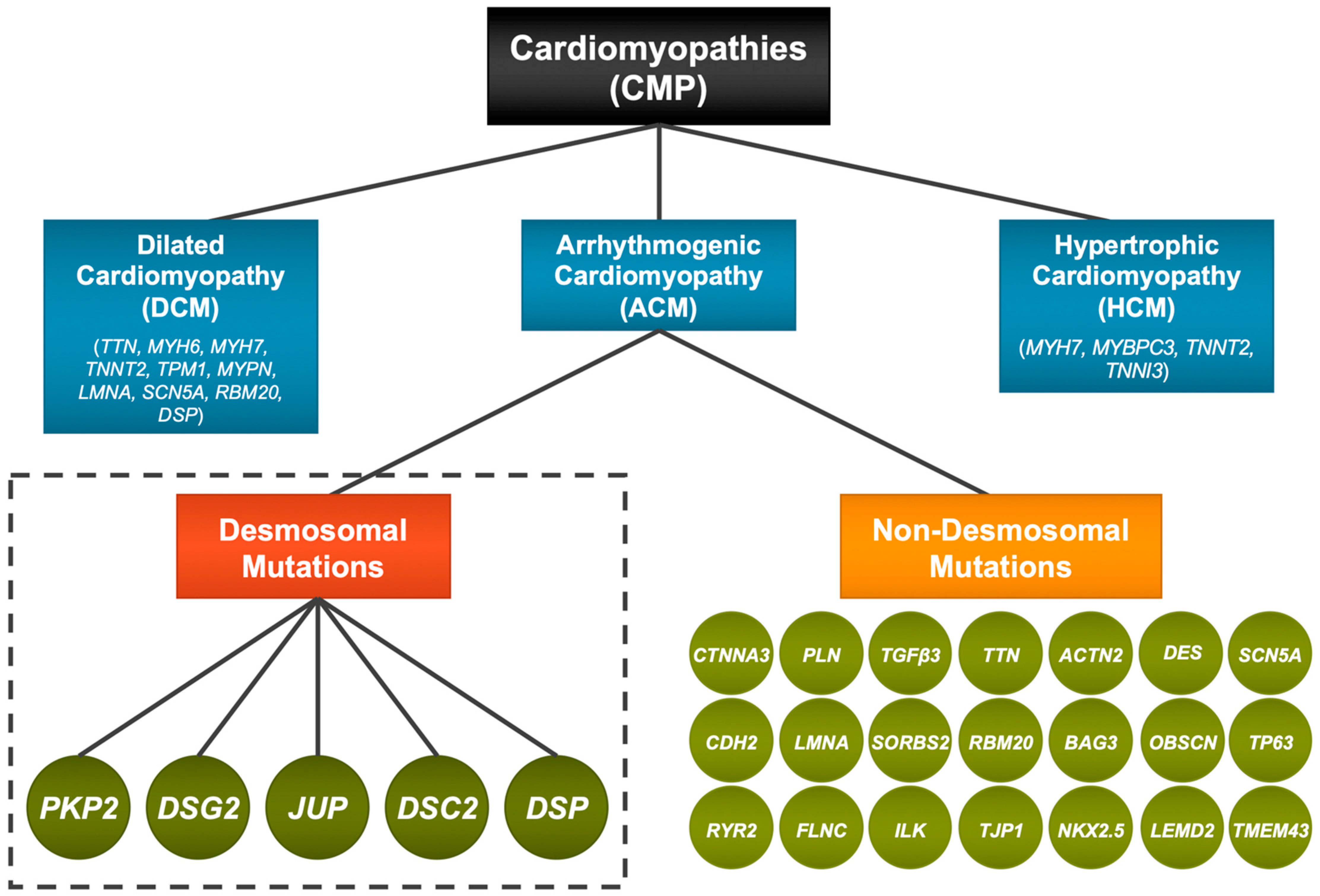
:1. Introduction—Cardiomyopathies and ACM

2. Clinical Phenotype
3. Desmosomal Proteins and Structures
4. Desmosomal Genes and ACM
4.1. Plakophilin (PKP)
4.2. Desmoglein (DSG)
4.3. Desmocollin (DSC)
4.4. Desmoplakin (DSP)
4.5. Plakoglobin (JUP)
4.6. Desmin
5. Human-Induced Pluripotent Stem Cells to Model dACM
5.1. PKP2 Mutant hiPSC Lines
5.2. DSG2 Mutant hiPSC Lines
5.3. DSP Mutant hiPSC Lines
5.4. DSC2 Mutant hiPSC Lines
5.5. Desmin Mutant hiPSC Lines
6. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Murray, B. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C): A Review of Molecular and Clinical Literature. J. Genet. Couns. 2012, 21, 494–504. [Google Scholar] [CrossRef]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pillichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and Digenic Heterozygosity Contributes to Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Protonotarios, N.; Tsatsopoulou, A. Naxos disease and Carvajal syndrome: Cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. 2004, 13, 185–194. [Google Scholar] [CrossRef]
- Gerull, B.; Kirchner, F.; Chong, J.X.; Tagoe, J.; Chandrasekharan, K.; Strohm, O.; Waggoner, D.; Ober, C.; Duff, H.J. Homozygous founder mutation in desmocollin-2 (dsc2) causes arrhythmogenic cardiomyopathy in the hutterite population. Circ. Cardiovasc. Genet. 2013, 6, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-?3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef]
- Good, J.-M.; Fellmann, F.; Bhuiyan, Z.A.; Rotman, S.; Pruvot, E.; Schläpfer, J. ACTN2 variant associated with a cardiac phenotype suggestive of left-dominant arrhythmogenic cardiomyopathy. Heart Case Rep. 2020, 6, 15–19. [Google Scholar] [CrossRef] [PubMed]
- van Tintelen, J.P.; Van Gelder, I.C.; Asimaki, A.; Suurmeijer, A.J.; Wiesfeld, A.C.; Jongbloed, J.D.; Wijngaard, A.v.D.; Kuks, J.B.; van Spaendonck-Zwarts, K.Y.; Notermans, N.; et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009, 6, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.J.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Rothenberg, E.; Sobreira, N.L.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.-C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 ( CDH2 ) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Alapi, K.Z.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef]
- Ding, Y.; Yang, J.; Chen, P.; Lu, T.; Jiao, K.; Tester, D.J.; Giudicessi, J.R.; Jiang, K.; Ackerman, M.J.; Li, Y.; et al. Knockout of SORBS2 Protein Disrupts the Structural Integrity of Intercalated Disc and Manifests Features of Arrhythmogenic Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e017055. [Google Scholar] [CrossRef]
- Parikh, V.N.; Caleshu, C.; Reuter, C.; Lazzeroni, L.C.; Ingles, J.; Garcia, J.; McCaleb, K.; Adesiyun, T.; Sedaghat-Hamedani, F.; Kumar, S.; et al. Regional Variation in RBM20 Causes a Highly Penetrant Arrhythmogenic Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005371. [Google Scholar] [CrossRef]
- Toro, R.; Pérez-Serra, A.; Campuzano, O.; Moncayo-Arlandi, J.; Allegue, C.; Iglesias, A.; Mangas, A.; Brugada, R. Familial Dilated Cardiomyopathy Caused by a Novel Frameshift in the BAG3 Gene. PLoS ONE 2016, 11, e0158730. [Google Scholar] [CrossRef]
- Chen, P.; Xiao, Y.; Wang, Y.; Zheng, Z.; Chen, L.; Yang, X.; Li, J.; Wu, W.; Zhang, S. Intracellular calcium current disorder and disease phenotype in OBSCN mutant iPSC-based cardiomyocytes in arrhythmogenic right ventricular cardiomyopathy. Theranostics 2020, 10, 11215–11229. [Google Scholar] [CrossRef]
- Poloni, G.; Calore, M.; Rigato, I.; Marras, E.; Minervini, G.; Mazzotti, E.; Lorenzon, A.; Mura, I.E.A.L.; Telatin, A.; Zara, I.; et al. A targeted next-generation gene panel reveals a novel heterozygous nonsense variant in the TP63 gene in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, 773–780. [Google Scholar] [CrossRef]
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001, 103, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Brun, F.; Gigli, M.; Graw, S.L.; Judge, D.P.; Merlo, M.; Murray, B.; Calkins, H.; Sinagra, G.; Taylor, M.R.; Mestroni, L.; et al. FLNC truncations cause arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2020, 57, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Rezazadeh, S.; Williams, T.; Munsie, N.M.; Liedtke, D.; Oh, T.; Ferrier, R.; Shen, Y.; Jones, S.J.; Stiegler, A.L.; et al. Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy. Transl. Res. 2019, 208, 15–29. [Google Scholar] [CrossRef]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-Exome Sequencing Identifies Pathogenic Variants in TJP1 Gene Associated With Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002123. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, N.; Chen, R.; Duff, H.J.; Seifer, C.M.; Buffo, I.; Huculak, C.; Clarke, S.; Clegg, R.; Jassal, D.S.; Gordon, P.M.; et al. Characterization of a Unique Form of Arrhythmic Cardiomyopathy Caused by Recessive Mutation in LEMD2. JACC Basic Transl. Sci. 2019, 4, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.-D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Brodehl, A. Genetic Animal Models for Arrhythmogenic Cardiomyopathy. Front. Physiol. 2020, 11, 624. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef]
- Sommariva, E.; Stadiotti, I.; Perrucci, G.L.; Tondo, C.; Pompilio, G. Cell models of arrhythmogenic cardiomyopathy: Advances and opportunities. Dis. Model. Mech. 2017, 10, 823–835. [Google Scholar] [CrossRef]
- Yang, D.; Gomez-Garcia, J.; Funakoshi, S.; Tran, T.; Fernandes, I.; Bader, G.D.; Laflamme, M.A.; Keller, G.M. Modeling human multi-lineage heart field development with pluripotent stem cells. Cell Stem Cell 2022, 29, 1382–1401.e8. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef] [PubMed]
- Sattar, Y.; Abdullah, H.M.; Samani, E.N.; Myla, M.; Ullah, W. Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An Updated Review of Diagnosis and Management. Cureus 2019, 11, e5381. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right Ventricular Dysplasia: A Report of 24 Adult Cases. Circulation 1982, 65, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Cerrone, M.; Saguner, A.M.; Brunckhorst, C.; Delmar, M.; Duru, F. Arrhythmogenic cardiomyopathy: An in-depth look at molecular mechanisms and clinical correlates. Trends Cardiovasc. Med. 2021, 31, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Moazzen, H.; Bolaji, M.D.; Leube, R.E. Desmosomes in Cell Fate Determination: From Cardiogenesis to Cardiomyopathy. Cells 2023, 12, 2122. [Google Scholar] [CrossRef]
- Gerçek, M.; Gerçek, M.; Kant, S.; Simsekyilmaz, S.; Kassner, A.; Milting, H.; Liehn, E.A.; Leube, R.E.; Krusche, C.A. Cardiomyocyte Hypertrophy in Arrhythmogenic Cardiomyopathy. Am. J. Pathol. 2017, 187, 752–766. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/Dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef]
- Pinamonti, B.; Brun, F.; Mestroni, L.; Sinagra, G. Arrhythmogenic right ventricular cardiomyopathy: From genetics to diagnostic and therapeutic challenges. World J. Cardiol. 2014, 6, 1234–1244. [Google Scholar] [CrossRef]
- James, C.A.; Jongbloed, J.D.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated With Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis. Med. 2021, 14, e003273. [Google Scholar] [CrossRef]
- Barrick, S.K.; Greenberg, M.J. Cardiac myosin contraction and mechanotransduction in health and disease. J. Biol. Chem. 2021, 297, 101297. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.F.; Mohler, P.J. Evolving form to fit function: Cardiomyocyte intercalated disc and transverse-tubule membranes. In Current Topics in Membranes; Academic Press Inc.: Cambridge, MA, USA, 2013; pp. 121–158. [Google Scholar] [CrossRef]
- Patel, D.M.; Green, K.J. Desmosomes in the heart: A review of clinical and mechanistic analyses. Cell Commun. Adhes. 2014, 21, 109–128. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Vermij, S.H.; Abriel, H.; Van Veen, T.A.B. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Biophys. Acta (BBA) Biomembr. 2008, 1778, 572–587. [Google Scholar] [CrossRef]
- Delva, E.; Tucker, D.K.; Kowalczyk, A.P. The Desmosome. Cold Spring Harb. Perspect. Biol. 2009, 1, a002543. [Google Scholar] [CrossRef]
- Harrison, O.J.; Brasch, J.; Lasso, G.; Katsamba, P.S.; Ahlsen, G.; Honig, B.; Shapiro, L. Structural basis of adhesive binding by desmocollins and desmogleins. Proc. Natl. Acad. Sci. USA 2016, 113, 7160–7165. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/ -catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef]
- Dubash, A.D.; Kam, C.Y.; Aguado, B.A.; Patel, D.M.; Delmar, M.; Shea, L.D.; Green, K.J. Plakophilin-2 loss promotes TGF-β1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J. Cell Biol. 2016, 212, 425–438. [Google Scholar] [CrossRef]
- Pérez-Hernández, M.; Marrón-Liñares, G.M.; Schlamp, F.; Heguy, A.; van Opbergen, C.J.M.; Mezzano, V.; Zhang, M.; Liang, F.-X.; Cerrone, M.; Delmar, M. Transcriptomic Coupling of PKP2 With Inflammatory and Immune Pathways Endogenous to Adult Cardiac Myocytes. Front. Physiol. 2021, 11, 1772. [Google Scholar] [CrossRef] [PubMed]
- Moncayo-Arlandi, J.; Brugada, R. Unmasking the molecular link between arrhythmogenic cardiomyopathy and Brugada syndrome. Nat. Rev. Cardiol. 2017, 14, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Ellesøe, S.G.; Johansen, M.M.; Bjerre, J.V.; Hjortdal, V.E.; Brunak, S.; Larsen, L.A. Familial Atrial Septal Defect and Sudden Cardiac Death: Identification of a NovelNKX2-5Mutation and a Review of the Literature. Congenit. Heart Dis. 2016, 11, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Tariq, H.; Bella, J.; Jowitt, T.A.; Holmes, D.F.; Rouhi, M.; Nie, Z.; Baldock, C.; Garrod, D.; Tabernero, L. Cadherin flexibility provides a key difference between desmosomes and adherens junctions. Proc. Natl. Acad. Sci. USA 2015, 112, 5395–5400. [Google Scholar] [CrossRef] [PubMed]
- Vreeker, A.; van Stuijvenberg, L.; Hund, T.J.; Mohler, P.J.; Nikkels, P.G.J.; van Veen, T.A.B. Assembly of the cardiac intercalated disk during pre- and postnatal development of the human heart. PLoS ONE 2014, 9, e94722. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Caspi, O.; Huber, I.; Gepstein, A.; Arbel, G.; Maizels, L.; Boulos, M.; Gepstein, L. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ. Cardiovasc. Genet. 2013, 6, 557–568. [Google Scholar] [CrossRef]
- Hawthorne, R.N.; Blazeski, A.; Lowenthal, J.; Kannan, S.; Teuben, R.; DiSilvestre, D.; Morrissette-McAlmon, J.; Saffitz, J.E.; Boheler, K.R.; James, C.A.; et al. Altered electrical, biomolecular, and immunologic phenotypes in a novel patient-derived stem cell model of Desmoglein-2 mutant ARVC. J. Clin. Med. 2021, 10, 3061. [Google Scholar] [CrossRef]
- Al-Jassar, C.; Bikker, H.; Overduin, M.; Chidgey, M. Mechanistic basis of desmosome-targeted diseases. J. Mol. Biol. 2013, 425, 4006–4022. [Google Scholar] [CrossRef]
- Meraviglia, V.; Alcalde, M.; Campuzano, O.; Bellin, M. Inflammation in the Pathogenesis of Arrhythmogenic Cardiomyopathy: Secondary Event or Active Driver? Front. Cardiovasc. Med. 2021, 8, 784715. [Google Scholar] [CrossRef]
- De Coster, T.; Claus, P.; Seemann, G.; Willems, R.; Sipido, K.R.; Panfilov, A.V. Myocyte remodeling due to fibro-fatty infiltrations influences arrhythmogenicity. Front. Physiol. 2018, 9, 1381. [Google Scholar] [CrossRef] [PubMed]
- Anumonwo, J.M.B.; Herron, T. Fatty Infiltration of the Myocardium and Arrhythmogenesis: Potential Cellular and Molecular Mechanisms. Front. Physiol. 2018, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Hoorntje, E.T.; Te Rijdt, W.P.T.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; Van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Fernández-Falgueras, A.; Iglesias, A.; Brugada, R. Brugada Syndrome and PKP2: Evidences and uncertainties. Int. J. Cardiol. 2016, 214, 403–405. [Google Scholar] [CrossRef]
- Yeh, Y.-T.; Hur, S.S.; Chang, J.; Wang, K.-C.; Chiu, J.-J.; Li, Y.-S.; Chien, S. Matrix Stiffness Regulates Endothelial Cell Proliferation through Septin 9. PLoS ONE 2012, 7, e46889. [Google Scholar] [CrossRef]
- Oxford, E.M.; Musa, H.; Maass, K.; Coombs, W.; Taffet, S.M.; Delmar, M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ. Res. 2007, 101, 703–711. [Google Scholar] [CrossRef]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.-N.; Jan, L.Y. Microtubule Plus-End-Tracking Proteins Target Gap Junctions Directly from the Cell Interior to Adherens Junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef]
- Sato, P.Y.; Coombs, W.; Lin, X.; Nekrasova, O.; Green, K.J.; Isom, L.L.; Taffet, S.M.; Delmar, M. Interactions between ankyrin-g, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ. Res. 2011, 109, 193–201. [Google Scholar] [CrossRef]
- Bonné, S.; Van Hengel, J.; Van Roy, F. Chromosomal Mapping of Human armadillo Genes Belonging to the p120 ctn /Plakophilin Subfamily. Genomics 1998, 51, 452–454. [Google Scholar] [CrossRef]
- Huber, A.H.; Nelson, W.; Weis, W.I. Three-Dimensional Structure of the Armadillo Repeat Region of β-Catenin. Cell 1997, 90, 871–882. [Google Scholar] [CrossRef]
- Loureiro, J.; Peifer, M. Roles of Armadillo, a Drosophila catenin, during central nervous system development. Curr. Biol. 1998, 8, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Delmar, M.; McKenna, W.J. The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circ. Res. 2010, 107, 700–714. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Charron, P.; Fressart, V.; de la Grandmaison, G.L.; Simon, F.; Gary, F.; Vite, A.; Hainque, B.; Hidden-Lucet, F.; Komajda, M.; et al. Plakophilin 2A is the dominant isoform in human heart tissue: Consequences for the genetic screening of arrhythmogenic right ventricular cardiomyopathy. Heart 2011, 97, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; A. MacRae, C.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, E.K.; Borowiec, K.; Franaszczyk, M.; Szperl, M.; Rampazzo, A.; Woźniak, O.; Roszczynko, M.; Śmigielski, W.; Lutyńska, A.; Hoffman, P. Pathogenic variants in plakophilin-2 gene (PKP2) are associated with better survival in arrhythmogenic right ventricular cardiomyopathy. J. Appl. Genet. 2021, 62, 613–620. [Google Scholar] [CrossRef]
- Syrris, P.; Ward, D.; Asimaki, A.; Sen-Chowdhry, S.; Ebrahim, H.Y.; Evans, A.; Hitomi, N.; Norman, M.; Pantazis, A.; Shaw, A.L.; et al. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 356–364. [Google Scholar] [CrossRef]
- Dalal, D.; James, C.; Devanagondi, R.; Tichnell, C.; Tucker, A.; Prakasa, K.; Spevak, P.J.; Bluemke, D.A.; Abraham, T.; Russell, S.D.; et al. Penetrance of Mutations in Plakophilin-2 Among Families With Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 1416–1424. [Google Scholar] [CrossRef]
- van Tintelen, J.P.; Entius, M.M.; Bhuiyan, Z.A.; Jongbloed, R.; Wiesfeld, A.C.; Wilde, A.A.; Boven, L.G.; Mannens, M.M.; van Langen, I.M.; Hofstra, R.M.; et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2006, 113, 1650–1658. [Google Scholar] [CrossRef]
- Leone, M.P.; Palumbo, P.; Saenen, J.; Mastroianno, S.; Castellana, S.; Amico, C.; Mazza, T.; Potenza, D.R.; Petracca, A.; Castori, M.; et al. Phenotypic Variability of a Pathogenic PKP2 Mutation in an Italian Family Affected by Arrhythmogenic Cardiomyopathy and Juvenile Sudden Death: Considerations From Molecular Autopsy to Sport Restriction. Front. Cardiovasc. Med. 2021, 8, 635141. [Google Scholar] [CrossRef]
- Mohammed, F.; Chidgey, M. Desmosomal protein structure and function and the impact of disease-causing mutations. J. Struct. Biol. 2021, 213, 107749. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Zhang, J.Z.; Itzhaki, I.; Zhang, S.L.; Chen, H.; Haddad, F.; Kitani, T.; Wilson, K.D.; Tian, L.; Shrestha, R.; et al. Determining the pathogenicity of a genomic variant of uncertain significance using CRISPR/Cas9 and human-induced pluripotent stem cells. Circulation 2018, 138, 2666–2681. [Google Scholar] [CrossRef] [PubMed]
- Khudiakov, A.; Zaytseva, A.; Perepelina, K.; Smolina, N.; Pervunina, T.; Vasichkina, E.; Karpushev, A.; Tomilin, A.; Malashicheva, A.; Kostareva, A. Sodium current abnormalities and deregulation of Wnt/β-catenin signaling in iPSC-derived cardiomyocytes generated from patient with arrhythmogenic cardiomyopathy harboring compound genetic variants in plakophilin 2 gene. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Cloonan, P.E.; Sundaram, S.; Liu, F.; Das, S.L.; Ewoldt, J.K.; Bays, J.L.; Tomp, S.; Toepfer, C.N.; Marsiglia, J.D.C.; et al. Plakophilin-2 truncating variants impair cardiac contractility by disrupting sarcomere stability and organization. Sci. Adv. 2021, 7, eabh3995. [Google Scholar] [CrossRef]
- Inoue, H.; Nakamura, S.; Higo, S.; Shiba, M.; Kohama, Y.; Kondo, T.; Kameda, S.; Tabata, T.; Okuno, S.; Ikeda, Y.; et al. Modeling reduced contractility and impaired desmosome assembly due to plakophilin-2 deficiency using isogenic iPS cell-derived cardiomyocytes. Stem Cell Rep. 2022, 17, 337–351. [Google Scholar] [CrossRef]
- Dorn, T.; Kornherr, J.; Parrotta, E.I.; Zawada, D.; Ayetey, H.; Santamaria, G.; Iop, L.; Mastantuono, E.; Sinnecker, D.; Goedel, A.; et al. Interplay of cell–cell contacts and RhoA/ MRTF—A signaling regulates cardiomyocyte identity. EMBO J. 2018, 37, e98133. [Google Scholar] [CrossRef]
- Moreau, A.; Reisqs, J.; Delanoe-Ayari, H.; Pierre, M.; Janin, A.; Deliniere, A.; Bessière, F.; Meli, A.C.; Charrabi, A.; Lafont, E.; et al. Deciphering DSC2 arrhythmogenic cardiomyopathy electrical instability: From ion channels to ECG and tailored drug therapy. Clin. Transl. Med. 2021, 11, e319. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.-H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. EP Eur. 2018, 20, f46–f56. [Google Scholar] [CrossRef]
- Shiba, M.; Higo, S.; Kondo, T.; Li, J.; Liu, L.; Ikeda, Y.; Kohama, Y.; Kameda, S.; Tabata, T.; Inoue, H.; et al. Phenotypic recapitulation and correction of desmoglein-2-deficient cardiomyopathy using human-induced pluripotent stem cell-derived cardiomyocytes. Hum. Mol. Genet. 2021, 30, 1384–1397. [Google Scholar] [CrossRef]
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient mutations linked to arrhythmogenic cardiomyopathy enhance calpain-mediated desmoplakin degradation. J. Clin. Investig. 2019, 4, e128643. [Google Scholar] [CrossRef]
- Gusev, K.; Khudiakov, A.; Zaytseva, A.; Perepelina, K.; Makeenok, S.; Kaznacheyeva, E.; Kostareva, A. Impact of the DSP-H1684R genetic variant on ion channels activity in iPSC-derived cardiomyocytes. Cell. Physiol. Biochem. 2020, 54, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Bliley, J.M.; Vermeer, M.C.S.C.; Duffy, R.M.; Batalov, I.; Kramer, D.; Tashman, J.W.; Shiwarski, D.J.; Lee, A.; Teplenin, A.S.; Volkers, L.; et al. Dynamic loading of human engineered heart tissue enhances contractile function and drives a desmosome-linked disease phenotype. Sci. Transl. Med. 2021, 13, eabd1817. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.-T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Arndt, A.K.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 2014, 6, 240ra74. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Protonotarios, A.; James, C.A.; Chelko, S.P.; Tichnell, C.; Murray, B.; Tsatsopoulou, A.; Anastasakis, A.; Riele, A.T.; Kléber, A.G.; et al. Characterizing the Molecular Pathology of Arrhythmogenic Cardiomyopathy in Patient Buccal Mucosa Cells. Circ. Arrhythmia Electrophysiol. 2016, 9, e003688. [Google Scholar] [CrossRef]
- Moriarty, M.A.; Ryan, R.; Lalor, P.; Dockery, P.; Byrnes, L.; Grealy, M. Loss of plakophilin 2 disrupts heart development in zebrafish. Int. J. Dev. Biol. 2012, 56, 711–718. [Google Scholar] [CrossRef]
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; García-Prieto, J.; García-Ruiz, J.M.; Pizarro, G.; Jiménez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruíz-Cabello, J.; et al. Exercise Triggers ARVC Phenotype in Mice Expressing a Disease-Causing Mutated Version of Human Plakophilin-2. J. Am. Coll. Cardiol. 2015, 65, 1438–1450. [Google Scholar] [CrossRef]
- Frank, J.; Cserhalmi-Friedman, P.B.; Ahmad, W.; Panteleyev, A.A.; Aita, V.M.; Christiano, A.M. Characterization of the desmosomal cadherin gene family: Genomic organization of two desmoglein genes on human chromosome 18q12. Exp. Dermatol. 2001, 10, 90–94. [Google Scholar] [CrossRef]
- Gehmlich, K.; Syrris, P.; Reimann, M.; Asimaki, A.; Ehler, E.; Evans, A.; Quarta, G.; Pantazis, A.; Saffitz, J.E.; McKenna, W.J. Molecular changes in the heart of a severe case of arrhythmogenic right ventricular cardiomyopathy caused by a desmoglein-2 null allele. Cardiovasc. Pathol. 2012, 21, 275–282. [Google Scholar] [CrossRef]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef]
- Fernandes, M.P.; Azevedo, O.; Pereira, V.; Calvo, L.; Lourenço, A. Arrhythmogenic right ventricular cardiomyopathy with left ventricular involvement: A novel splice site mutation in the DSG2 gene. Cardiology 2015, 130, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Lao, N.; Laiq, Z.; Courson, J.; Al-Quthami, A. Left-dominant arrhythmogenic cardiomyopathy: An association with desmoglein-2 gene mutation—A case report. Eur. Hear. J. Case Rep. 2021, 5, ytab213. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Meshkov, A.; Myasnikov, R.; Kiseleva, A.; Kulikova, O.; Klauke, B.; Sotnikova, E.; Stanasiuk, C.; Divashuk, M.; Pohl, G.M.; et al. Hemi- and homozygous loss-of-function mutations in dsg2 (Desmoglein-2) cause recessive arrhythmogenic cardiomyopathy with an early onset. Int. J. Mol. Sci. 2021, 22, 3786. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Li, W.; Zhang, Q.; Zhou, L.; Hua, Y.; Duan, H.; Li, Y. Misdiagnosed myocarditis in arrhythmogenic cardiomyopathy induced by a homozygous variant of DSG2: A case report. Front. Cardiovasc. Med. 2023, 10, 1150657. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.E.; Delaney, P.J.; Thenet, D.; Murtough, S.; Webb, C.M.; Zaman, N.; Tsisanova, E.; Mastroianni, G.; Walker, S.L.M.; Westaby, J.D.; et al. Early inflammation precedes cardiac fibrosis and heart failure in desmoglein 2 murine model of arrhythmogenic cardiomyopathy. Cell Tissue Res. 2021, 386, 79–98. [Google Scholar] [CrossRef]
- Dieding, M.; Debus, J.D.; Kerkhoff, R.; Gaertner-Rommel, A.; Walhorn, V.; Milting, H.; Anselmetti, D. Arrhythmogenic cardiomyopathy related DSG2 mutations affect desmosomal cadherin binding kinetics. Sci. Rep. 2017, 7, 13791. [Google Scholar] [CrossRef]
- Hermida, A.; Fressart, V.; Hidden-Lucet, F.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; et al. High risk of heart failure associated with desmoglein-2 mutations compared to plakophilin-2 mutations in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Heart Fail. 2019, 21, 792–800. [Google Scholar] [CrossRef]
- Eshkind, L.; Tian, Q.; Schmidt, A.; Franke, W.W.; Windoffer, R.; Leube, R.E. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur. J. Cell Biol. 2002, 81, 592–598. [Google Scholar] [CrossRef]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; Hoff, M.J.v.D.; de Bakker, J.M.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef]
- Krusche, C.A.; Holthöfer, B.; Hofe, V.; van de Sandt, A.M.; Eshkind, L.; Bockamp, E.; Merx, M.W.; Kant, S.; Windoffer, R.; Leube, R.E. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res. Cardiol. 2011, 106, 617–633. [Google Scholar] [CrossRef]
- Rizzo, S.; Lodder, E.M.; Verkerk, A.O.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.R. Intercalated disc abnormalities, reduced Na+ current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 2012, 95, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Holthöfer, B.; Magin, T.M.; Krusche, C.A.; Leube, R.E. Desmoglein 2–Dependent Arrhythmogenic Cardiomyopathy Is Caused by a Loss of Adhesive Function. Circ. Cardiovasc. Genet. 2015, 8, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. J. Clin. Investig. 2016, 1, e85923. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, R.; Huang, Y.; Yang, Z.; Xian, J.; Huang, J.; Qiu, Z.; Lin, X.; Zhang, M.; Chen, H.; et al. Reactivation of PPARα alleviates myocardial lipid accumulation and cardiac dysfunction by improving fatty acid β-oxidation in Dsg2-deficient arrhythmogenic cardiomyopathy. Acta Pharm. Sin. B 2023, 13, 192–203. [Google Scholar] [CrossRef]
- Qiu, Z.; Zhao, Y.; Tao, T.; Guo, W.; Liu, R.; Huang, J.; Xu, G. Activation of PPARα Ameliorates Cardiac Fibrosis in Dsg2-Deficient Arrhythmogenic Cardiomyopathy. Cells 2022, 11, 3184. [Google Scholar] [CrossRef]
- Hunt, D.M.; Sahota, V.K.; Taylor, K.; Šimrak, D.; Hornigold, N.; Arnemann, J.; Wolfe, J.; Buxton, R.S. Clustered Cadherin Genes: A Sequence-Ready Contig for the Desmosomal Cadherin Locus on Human Chromosome 18. Genomics 1999, 62, 445–455. [Google Scholar] [CrossRef]
- Green, K.J.; Gaudry, C.A. Are desmosomes more than tethers for intermediate filaments? Nat. Rev. Mol. Cell Biol. 2000, 1, 208–216. [Google Scholar] [CrossRef]
- James, C.A.; Calkins, H. Annual Review of Medicine Arrhythmogenic Right Ventricular Cardiomyopathy: Progress Toward Personalized Management. Annu. Rev. Med. 2019, 70, 1–18. [Google Scholar] [CrossRef]
- Gehmlich, K.; Syrris, P.; Peskett, E.; Evans, A.; Ehler, E.; Asimaki, A.; Anastasakis, A.; Tsatsopoulou, A.; Vouliotis, A.-I.; Stefanadis, C.; et al. Mechanistic insights into arrhythmogenic right ventricular cardiomyopathy caused by desmocollin-2 mutations. Cardiovasc. Res. 2011, 90, 77–87. [Google Scholar] [CrossRef]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gärtner, A.; et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J. Mol. Cell. Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.-X.; Gordon, P.M.K.; Nygren, A.; et al. Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Yamamoto, T.; Nakamura, Y.; Sufu-Shimizu, Y.; Nanno, T.; Fukuda, M.; Ono, M.; Oda, T.; Okuda, S.; Ueyama, T.; et al. G790del mutation in DSC2 alone is insufficient to develop the pathogenesis of ARVC in a mouse model. Biochem. Biophys. Rep. 2020, 21, 100711. [Google Scholar] [CrossRef] [PubMed]
- Pohl, G.M.; Göz, M.; Gaertner, A.; Brodehl, A.; Cimen, T.; Saguner, A.M.; Schulze-Bahr, E.; Walhorn, V.; Anselmetti, D.; Milting, H. Cardiomyopathy related desmocollin-2 prodomain variants affect the intracellular cadherin transport and processing. Front. Cardiovasc. Med. 2023, 10, 1127261. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-Y.; Cheng, L.-T.; Wang, Z.-F.; Wu, Y.-Q. Desmoplakin and clinical manifestations of desmoplakin cardiomyopathy. Chin. Med. J. 2021, 134, 1771–1779. [Google Scholar] [CrossRef]
- Kaplan, S.R.; Gard, J.J.; Carvajal-Huerta, L.; Ruiz-Cabezas, J.C.; Thiene, G.; Saffitz, J.E. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc. Pathol. 2004, 13, 26–32. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Report Mutation in Human Desmoplakin Domain Binding to Plakoglobin Causes a Dominant Form of Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Prasad, S.K.; Syrris, P.; Wage, R.; Ward, D.; Merrifield, R.; Smith, G.C.; Firmin, D.N.; Pennell, D.J.; McKenna, W.J. Cardiovascular Magnetic Resonance in Arrhythmogenic Right Ventricular Cardiomyopathy Revisited: Comparison With Task Force Criteria and Genotype. J. Am. Coll. Cardiol. 2006, 48, 2132–2140. [Google Scholar] [CrossRef]
- Gallicano, G.I.; Kouklis, P.; Bauer, C.; Yin, M.; Vasioukhin, V.; Degenstein, L.; Fuchs, E. Desmoplakin Is Required Early in Development for Assembly of Desmosomes and Cytoskeletal Linkage. J. Cell Biol. 1998, 143, 2009–2022. [Google Scholar] [CrossRef]
- Huang, X.; Yan, L.; Kou, S.; Meng, J.; Lu, Z.; Lin, C.-P.; Liu, C.; Zhang, H. Generation and characterization of a Myh6-driven Cre knockin mouse line. Transgenic Res. 2021, 30, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.L.; Manring, H.R.; Wallace, M.J.; Argall, A.; Dew, T.; Papaioannou, P.; Antwi-Boasiako, S.; Xu, X.; Campbell, S.G.; Akar, F.G.; et al. Humanized Dsp ACM Mouse Model Displays Stress-Induced Cardiac Electrical and Structural Phenotypes. Cells 2022, 11, 3049. [Google Scholar] [CrossRef] [PubMed]
- Pratt, C.H.; Potter, C.S.; Fairfield, H.; Reinholdt, L.G.; Bergstrom, D.E.; Harris, B.S.; Greenstein, I.; Dadras, S.S.; Liang, B.T.; Schofield, P.N.; et al. Dsprul: A spontaneous mouse mutation in desmoplakin as a model of Carvajal-Huerta syndrome. Exp. Mol. Pathol. 2015, 98, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.; Brinkmann, V.; Ledermann, B.; Behrend, M.; Grund, C.; Thalhammer, C.; Vogel, F.; Birchmeier, C.; Günthert, U.; Franke, W.W.; et al. Targeted Mutation of Plakoglobin in Mice Reveals Essential Functions of Desmosomes in the Embryonic Heart. J. Cell Biol. 1996, 135, 215–225. [Google Scholar] [CrossRef]
- Lewis, J.E.; Wahl, J.K.; Sass, K.M.; Jensen, P.J.; Johnson, K.R.; Wheelock, M.J. Cross-Talk between Adherens Junctions and Desmosomes Depends on Plakoglobin. J. Cell Biol. 1997, 136, 919–934. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, C.; Li, Y.; Yu, R. Whole-exome sequencing identified a de novo mutation of junction plakoglobin (p.r577c) in a chinese patient with arrhythmogenic right ventricular cardiomyopathyy. BioMed Res. Int. 2019, 2019, 9103860. [Google Scholar] [CrossRef]
- Kirchhof, P.; Fabritz, L.; Zwiener, M.; Witt, H.; Schäfers, M.; Zellerhoff, S.; Paul, M.; Athai, T.; Hiller, K.-H.; Baba, H.A.; et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 2006, 114, 1799–1806. [Google Scholar] [CrossRef]
- Fabritz, L.; Hoogendijk, M.G.; Scicluna, B.P.; van Amersfoorth, S.C.; Fortmueller, L.; Wolf, S.; Laakmann, S.; Kreienkamp, N.; Piccini, I.; Breithardt, G.; et al. Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-deficient mice. J. Am. Coll. Cardiol. 2011, 57, 740–750. [Google Scholar] [CrossRef]
- Lombardi, R.; da Graca Cabreira-Hansen, M.; Bell, A.; Fromm, R.R.; Willerson, J.T.; Marian, A. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2011, 109, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.-F.; Haneline, L.S.; Field, L.J.; et al. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, A.; Beffagna, G.; Bauce, B.; De Bortoli, M.; Mura, I.E.L.; Calore, M.; Dazzo, E.; Basso, C.; Nava, A.; Thiene, G.; et al. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am. J. Cardiol. 2013, 111, 400–405. [Google Scholar] [CrossRef]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Thiene, G.; Basso, C.; Pilichou, K.; Marinas, M.B. Desmosomal Arrhythmogenic Cardiomyopathy: The Story Telling of a Genetically Determined Heart Muscle Disease. Biomedicines 2023, 11, 2018. [Google Scholar] [CrossRef]
- Maggi, L.; Mavroidis, M.; Psarras, S.; Capetanaki, Y.; Lattanzi, G. Skeletal and Cardiac Muscle Disorders Caused by Mutations in Genes Encoding Intermediate Filament Proteins. Int. J. Mol. Sci. 2021, 22, 4256. [Google Scholar] [CrossRef]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Lammerding, J. Connecting the plasma membrane to the nucleus by intermediate filaments. Mol. Biol. Cell 2017, 28, 695–696. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Cytoplasmic Intermediate Filaments in Cell Biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef]
- Hol, E.M.; Capetanaki, Y. Type III Intermediate Filaments Desmin, Glial Fibrillary Acidic Protein (GFAP), Vimentin, and Peripherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a021642. [Google Scholar] [CrossRef]
- Wahbi, K.; Béhin, A.; Charron, P.; Dunand, M.; Richard, P.; Meune, C.; Vicart, P.; Laforêt, P.; Stojkovic, T.; Bécane, H.M.; et al. High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: A 10-year longitudinal study. Neuromuscul. Disord. 2012, 22, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Psarras, S.; Mavroidis, M.; Sanoudou, D.; Davos, C.H.; Xanthou, G.; Varela, A.E.; Panoutsakopoulou, V.; Capetanaki, Y. Regulation of adverse remodelling by osteopontin in a genetic heart failure model. Eur. Heart J. 2011, 33, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Rapti, K.; Diokmetzidou, A.; Kloukina, I.; Milner, D.J.; Varela, A.; Davos, C.H.; Capetanaki, Y. Opposite effects of catalase and MnSOD ectopic expression on stress induced defects and mortality in the desmin deficient cardiomyopathy model. Free. Radic. Biol. Med. 2017, 110, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Boukens, B.J.; Rivaud, M.R.; Rentschler, S.; Coronel, R. Misinterpretation of the mouse ECG: ‘musing the waves of Mus musculus’. J. Physiol. 2014, 592, 4613–4626. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement from the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, C.W.; Okawa, S.; Chuva de Sousa Lopes, S.M.; van Iperen, L.; Passier, R.; Braam, S.R.; Tertoolen, L.G.; del Sol, A.; Davis, R.P.; Mummery, C.L. Transcriptome of human foetal heart compared with cardiomyocytes from pluripotent stem cells. Development 2015, 142, 3231–3238. [Google Scholar] [CrossRef]
- Fatima, A.; Xu, G.; Shao, K.; Papadopoulos, S.; Lehmann, M.; Arnáiz-Cot, J.J.; Rosa, A.O.; Nguemo, F.; Matzkies, M.; Dittmann, S.; et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell. Physiol. Biochem. 2011, 28, 579–592. [Google Scholar] [CrossRef]
- Jung, C.B.; Moretti, A.; Schnitzler, M.M.Y.; Iop, L.; Storch, U.; Bellin, M.; Dorn, T.; Ruppenthal, S.; Pfeiffer, S.; Goedel, A.; et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol. Med. 2012, 4, 180–191. [Google Scholar] [CrossRef]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef]
- Kinnear, C.; Chang, W.Y.; Khattak, S.; Hinek, A.; Thompson, T.; Rodrigues, D.d.C.; Kennedy, K.; Mahmut, N.; Pasceri, P.; Stanford, W.L.; et al. Modeling and Rescue of the Vascular Phenotype of Williams-Beuren Syndrome in Patient Induced Pluripotent Stem Cells. Stem Cells Transl. Med. 2013, 2, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Shao, N.-Y.; Sa, S.; Li, D.; Termglinchan, V.; Ameen, M.; Karakikes, I.; Sosa, G.; Grubert, F.; Lee, J.; et al. Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20, 490–504.e5. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Cayo, M.A.; Cai, J.; DeLaForest, A.; Noto, F.K.; Nagaoka, M.; Clark, B.S.; Collery, R.F.; Si-Tayeb, K.; Duncan, S.A. JD induced pluripotent stem cell-derived hepatocytes faithfully recapitulate the pathophysiology of familial hypercholesterolemia. Hepatology 2012, 56, 2163–2171. [Google Scholar] [CrossRef]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra47. [Google Scholar] [CrossRef]
- Brodehl, A.; Ebbinghaus, H.; Deutsch, M.-A.; Gummert, J.; Gärtner, A.; Ratnavadivel, S.; Milting, H. Human induced pluripotent stem-cell-derived cardiomyocytes as models for genetic cardiomyopathies. Int. J. Mol. Sci. 2019, 20, 4381. [Google Scholar] [CrossRef]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome Editing in Induced Pluripotent Stem Cells using CRISPR/Cas9. Stem Cell Rev. Rep. 2018, 14, 323–336. [Google Scholar] [CrossRef]
- Zhang, J.; Chou, O.H.-I.; Tse, Y.-L.; Ng, K.-M.; Tse, H.-F. Application of patient-specific ipscs for modelling and treatment of x-linked cardiomyopathies. Int. J. Mol. Sci. 2021, 22, 8132. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Carrier, L. Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes—A systematic review. Pflügers Arch. -Eur. J. Physiol. 2019, 471, 755–768. [Google Scholar] [CrossRef]
- Bellin, M.; Casini, S.; Davis, R.P.; D’Aniello, C.; Haas, J.; Oostwaard, D.W.-V.; Tertoolen, L.G.J.; Jung, C.B.; Elliott, D.A.; Welling, A.; et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013, 32, 3161–3175. [Google Scholar] [CrossRef]
- Ma, D.; Wei, H.; Lu, J.; Ho, S.; Zhang, G.; Sun, X.; Oh, Y.; Tan, S.H.; Ng, M.L.; Shim, W.; et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 1122–1133. [Google Scholar] [CrossRef]
- Akdis, D.; Saguner, A.M.; Shah, K.; Wei, C.; Medeiros-Domingo, A.; von Eckardstein, A.; Lüscher, T.F.; Brunckhorst, C.; Chen, H.V.; Duru, F. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: From a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur. Heart J. 2017, 38, 1498–1508. [Google Scholar] [CrossRef]
- Awad, M.M.; Dalal, D.; Tichnell, C.; James, C.; Tucker, A.; Abraham, T.; Spevak, P.J.; Calkins, H.; Judge, D.P. Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum. Mutat. 2006, 27, 1157. [Google Scholar] [CrossRef]
- Dalal, D.; Molin, L.H.; Piccini, J.; Tichnell, C.; James, C.; Bomma, C.; Prakasa, K.; Towbin, J.A.; Marcus, F.I.; Spevak, P.J.; et al. Clinical features of arrhythmogenic right ventricular dysplasia/ cardiomyopathy associated with mutations in plakophilin-2. Circulation 2006, 113, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.-Y.; Wei, C.-Y.; Shah, K.; Wong, J.; Wang, C.; Chen, H.-S.V. Maturation-Based Model of Arrhythmogenic Right Ventricular Dysplasia Using Patient-Specific Induced Pluripotent Stem Cells. Circ. J. 2015, 79, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Martewicz, S.; Luni, C.; Serena, E.; Pavan, P.; Chen, H.-S.V.; Rampazzo, A.; Elvassore, N. Transcriptomic Characterization of a Human In Vitro Model of Arrhythmogenic Cardiomyopathy Under Topological and Mechanical Stimuli. Ann. Biomed. Eng. 2019, 47, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Blazeski, A.; Lowenthal, J.; Wang, Y.; Teuben, R.; Zhu, R.; Gerecht, S.; Tomaselli, G.; Tung, L. Engineered Heart Slice Model of Arrhythmogenic Cardiomyopathy Using Plakophilin-2 Mutant Myocytes. Tissue Eng. Part A 2019, 25, 725–735. [Google Scholar] [CrossRef]
- Blazeski, A.; Lowenthal, J.; Zhu, R.; Ewoldt, J.; Boheler, K.R.; Tung, L. Functional Properties of Engineered Heart Slices Incorporating Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cell Rep. 2019, 12, 982–995. [Google Scholar] [CrossRef]
- Chelko, S.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
- Buljubasic, F.; El-Battrawy, I.; Lan, H.; Lomada, S.K.; Chatterjee, A.; Zhao, Z.; Li, X.; Zhong, R.; Xu, Q.; Huang, M.; et al. Nucleoside diphosphate kinase B contributes to arrhythmogenesis in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with arrhythmogenic right ventricular cardiomyopathy. J. Clin. Med. 2020, 9, 486. [Google Scholar] [CrossRef] [PubMed]
- Weisbrod, D.; Peretz, A.; Ziskind, A.; Menaker, N.; Oz, S.; Barad, L.; Eliyahu, S.; Itskovitz-Eldor, J.; Dascal, N.; Khananshvili, D.; et al. SK4 Ca2+ activated K + channel is a critical player in cardiac pacemaker derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1685–E1694. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Li, Z.; Ko, K.; Choudhury, P.; Albaqumi, M.; Johnson, A.K.; Yan, Y.; Backer, J.M.; Unutmaz, D.; Coetzee, W.A.; et al. Histidine Phosphorylation of the Potassium Channel KCa3.1 by Nucleoside Diphosphate Kinase B Is Required for Activation of KCa3.1 and CD4 T Cells. Mol. Cell 2006, 24, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Reisqs, J.; Moreau, A.; Charrabi, A.; Sleiman, Y.; Meli, A.C.; Millat, G.; Briand, V.; Beauverger, P.; Richard, S.; Chevalier, P. The PPARγ pathway determines electrophysiological remodelling and arrhythmia risks in DSC2 arrhythmogenic cardiomyopathy. Clin. Transl. Med. 2022, 12, e748. [Google Scholar] [CrossRef] [PubMed]
- Reisqs, J.-B.; Moreau, A.; Sleiman, Y.; Charrabi, A.; Delinière, A.; Bessière, F.; Gardey, K.; Richard, S.; Chevalier, P. Spironolactone as a Potential New Treatment to Prevent Arrhythmias in Arrhythmogenic Cardiomyopathy Cell Model. J. Pers. Med. 2023, 13, 335. [Google Scholar] [CrossRef]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The Novel Desmin Variant p.Leu115Ile Is Associated With a Unique Form of Biventricular Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2021, 37, 857–866. [Google Scholar] [CrossRef]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive cardiomyopathy is caused by a novel homozygous desmin (DES) mutation p.Y122H leading to a severe filament assembly defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef]
- Khudiakov, A.; Kostina, D.; Zlotina, A.; Nikulina, T.; Sergushichev, A.; Gudkova, A.; Tomilin, A.; Malashicheva, A.; Kostareva, A. Generation of iPSC line from desmin-related cardiomyopathy patient carrying splice site mutation of DES gene. Stem Cell Res. 2017, 24, 77–80. [Google Scholar] [CrossRef]
- Shanks, N.; Greek, R.; Greek, J. Are animal models predictive for humans? Philos. Ethic. Humanit. Med. 2009, 4, 2. [Google Scholar] [CrossRef]
- Kilpinen, H.; Goncalves, A.; Leha, A.; Afzal, V.; Alasoo, K.; Ashford, S.; Bala, S.; Bensaddek, D.; Casale, F.P.; Culley, O.J.; et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 2017, 546, 370–375. [Google Scholar] [CrossRef]
- Luecke, L.B.; Waas, M.; Littrell, J.; Wojtkiewicz, M.; Castro, C.; Burkovetskaya, M.; Schuette, E.N.; Buchberger, A.R.; Churko, J.M.; Chalise, U.; et al. Surfaceome mapping of primary human heart cells with CellSurfer uncovers cardiomyocyte surface protein LSMEM2 and proteome dynamics in failing hearts. Nat. Cardiovasc. Res. 2023, 2, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2012, 12, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.A.; Chen, E.Z.; Tung, L.; Boheler, K.R.; Kwon, C. Maturing heart muscle cells: Mechanisms and transcriptomic insights. Semin. Cell Dev. Biol. 2021, 119, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Poon, E.N.-Y.; Luo, X.-L.; Webb, S.E.; Yan, B.; Zhao, R.; Wu, S.C.M.; Yang, Y.; Zhang, P.; Bai, H.; Shao, J.; et al. The cell surface marker CD36 selectively identifies matured, mitochondria-rich hPSC-cardiomyocytes. Cell Res. 2020, 30, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C., Jr.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Zhao, Y.; Li, H.; Yang, Y.; Chen, M.; Wang, X.; Luo, M.; Wang, K. Engineering the maturation of stem cell-derived cardiomyocytes. Front. Bioeng. Biotechnol. 2023, 11, 1155052. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef]
- Lyra-Leite, D.M.; Gutiérrez-Gutiérrez, O.; Wang, M.; Zhou, Y.; Cyganek, L.; Burridge, P.W. A review of protocols for human iPSC culture, cardiac differentiation, subtype-specification, maturation, and direct reprogramming. STAR Protoc. 2022, 3, 101560. [Google Scholar] [CrossRef]
- Wu, P.; Deng, G.; Sai, X.; Guo, H.; Huang, H.; Zhu, P. Maturation strategies and limitations of induced pluripotent stem cell-derived cardiomyocytes. Biosci. Rep. 2021, 41, BSR20200833. [Google Scholar] [CrossRef]
- Jiang, Y.; Park, P.; Hong, S.-M.; Ban, K. Maturation of cardiomyocytes derived from human pluripotent stem cells: Current strategies and limitations. Mol. Cells 2018, 41, 613–621. [Google Scholar] [CrossRef]
- Thomas, D.; Cunningham, N.J.; Shenoy, S.; Wu, J.C. Human-induced pluripotent stem cells in cardiovascular research: Current approaches in cardiac differentiation, maturation strategies, and scalable production. Cardiovasc. Res. 2022, 118, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, B.; Asimaki, A.; Landstrom, A.P.; Khanji, M.Y.; Odening, K.E.; Cooper, L.T.; Marchlinski, F.E.; Gelzer, A.R.; Semsarian, C.; Reichlin, T.; et al. Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review. Circulation 2021, 144, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Scheel, P.J.; Murray, B.; Tichnell, C.; James, C.A.; Tandri, H.; Calkins, H.; Chelko, S.P.; Gilotra, N.A. Arrhythmogenic Right Ventricular Cardiomyopathy Presenting as Clinical Myocarditis in Women. Am. J. Cardiol. 2021, 145, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lowenthal, J.; Tomaselli, G.F.; Tung, L. Human iPSC models of cardiac electrophysiology and arrhythmia. In iPSCs—State of the Science; Elsevier: Amsterdam, The Netherlands, 2022; pp. 29–93. [Google Scholar] [CrossRef]
- Redfern, W.; Carlsson, L.; Davis, A.; Lynch, W.; MacKenzie, I.; Palethorpe, S.; Siegl, P.; Strang, I.; Sullivan, A.; Wallis, R.; et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Blinova, K.; Dang, Q.; Millard, D.; Smith, G.; Pierson, J.; Guo, L.; Brock, M.; Lu, H.R.; Kraushaar, U.; Zeng, H.; et al. International Multisite Study of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Drug Proarrhythmic Potential Assessment. Cell Rep. 2018, 24, 3582–3592. [Google Scholar] [CrossRef]
- Kelly, M.I.; Albahrani, M.; Castro, C.; Poon, E.; Yan, B.; Littrell, J.; Waas, M.; Boheler, K.R.; Gundry, R.L. Importance of evaluating protein glycosylation in pluripotent stem cell-derived cardiomyocytes for research and clinical applications. Pflug. Arch. Eur. J. Physiol. 2021, 473, 1041–1059. [Google Scholar] [CrossRef]
- Brodehl, A.; Stanasiuk, C.; Anselmetti, D.; Gummert, J.; Milting, H. Incorporation of desmocollin-2 into the plasma membrane requires N-glycosylation at multiple sites. FEBS Open Bio 2019, 9, 996–1007. [Google Scholar] [CrossRef]
- Gehmlich, K.; Asimaki, A.; Cahill, T.J.; Ehler, E.; Syrris, P.; Zachara, E.; Re, F.; Avella, A.; Monserrat, L.; Saffitz, J.E.; et al. Novel missense mutations in exon 15 of desmoglein-2: Role of the intracellular cadherin segment in arrhythmogenic right ventricular cardiomyopathy? Heart Rhythm 2010, 7, 1446–1453. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation | T/M | Pkp2 | Pkg | Dsg2 | Dsp | Dsc2 | N-cad | Cx43 | Nav1.5 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PKP2 | p.L614P | M | ▼▽ | ▼▽ | ■□ | ■□ | ■□ | [173] | |||
| p.G828LfsX835 | T | ▼ | ▼ | ▽ | [28] | ||||||
| p.K672RfsX683 | T | ▼▽ | ▼ | ||||||||
| p.A324fsX355 | T | ▼▽ | ▼□ | □ | ▼ | [58] | |||||
| p.D50SfsX110 | T | ▼▽ | ▼□ | □ | ▼ | ||||||
| p.V587AfsX655 | T | ▼▼□ | ▼ | [87] | |||||||
| p.A324fsX355 | T | ▽ | ▼▼▽ | ▽ | ▽ | ▼▽ | ▼▽ | [179] | |||
| p.G828LfsX835 | T | ▼□ | ▼□ | ▽ | ▼▽ | [178] | |||||
| p.Y119MfsX141 + p.K859R | T+M | ▼▼ | ▼■ | ■■□ | [84] | ||||||
| p.D109AfsX119 * | T | ▼▼▽ | ▼ | ■ | ▼ | [85] | |||||
| p.D109AfsX119 * | T | ▼▼▽ | ▼ | ■ | ▼ | ||||||
| p.D410fsX425 | T | ▼▼▽ | ■■ | ■■ | ■ | ■■ | ■■ | ■■ | [86] | ||
| p.D410fsX412 * | T | ▼▼▽ | ▼▼ | ▼▼ | ▼ | ▼▼ | ▼▼ | ■■ | |||
| DSG2 | p.G638R† | M | NA | ▽ | [89] | ||||||
| p.D787MfsX807 | T | ■■▽ | ■■□ | ▼▼▽ | □ | △ | [59] | ||||
| p.R119X | M | ■■ | ■■ | ▼▼▽ | ■■ | ■ | ■ | [90] | |||
| DSP | p.R451G | M | ▼ | ▼ | [91] | ||||||
| p.H1684R | M | ■ | ■■ | [92] | |||||||
| c.273+5G>A + p.R2229X2261 | T | ▼▼▽ | ▼ | [93] | |||||||
| DSC2 | p.R132C | M | ▽ | ▼▽ | [88] | ||||||
| Heterozygous mutation (not shaded) | ▼ decreased relative to control | ||||||||||
| Compound heterozygous mutation (shaded red) | ■ no change relative to control | ||||||||||
| Homozygous mutation (shaded blue) | ▲ increased relative to control | ||||||||||
| Mutant gene product (shaded gray) | ▲■▼ membrane protein (immunofluoresence) | ||||||||||
| * Artificially introduced mutation (not patient derived) | ▲■▼ total protein abundance (Western blot) | ||||||||||
| † Assumed heterozygous, although not specified | △□▽ total mRNA abundance (qPCR / RNA-seq) | ||||||||||
| Molecular Data | Single Channel Currents | Excitation, Conduction, & Synchrony | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Mutation | SCN5A/Nav1.5 | GJA/Cx43 | SK3/KCa2.3 | SK4/KCa3.1 | KCNJ11/Kir6.2 | KCNQ1/Kv7.1 | KCNJ2/Kir2.1 | KCNH2/Kv11.1 | SLC8A1/NCX1 | INa | ICaL | INCX | Ito | IK | Ikr | Iks | ISK | ISK4 | IKATP | AP probability | Resting potential | AP upstroke velocity | AP upstroke heterogeneity | AP amplitude | AP duration | AP frequency | Conduction velocity | Arrhythmic events | Refs. |
| PKP2 | p.G828LfsX835 | ▽ | ▼ | ■ | ▼ | ▼ | ■ | [28,68] | ||||||||||||||||||||||
| p.A324fsX355 | ▼ | ▼ | [58] | |||||||||||||||||||||||||||
| p.Y119MfsX141 + p.K859R | ■■□ | ▼ | ▼ | ■ | [84] | |||||||||||||||||||||||||
| p.D109AfsX119 * | ▼ | ▲ | [85] | |||||||||||||||||||||||||||
| p.D109AfsX119 * | ▼ | ▲ | ||||||||||||||||||||||||||||
| DSG2 | p.G638R † | ▽ | ▽ | ▲△ | ▽ | ▼ | ■ | ▼ | ▼ | ▲ | ■ | ▼ | ▲ | ▼ | ■ | ▼ | ▼ | ■ | ▲ | ▲ | [89,182] | |||||||||
| p.D787MfsX807 | △ | □ | △ | □ | □ | ■ | ▲ | ▼ | ■ | [59] | ||||||||||||||||||||
| p.R119X | ▼ | ▲ | [90] | |||||||||||||||||||||||||||
| DSP | p.R451G | ▼ | ■ | [91] | ||||||||||||||||||||||||||
| p.H1684R | ▼ | ▼ | ▲ | ▼ | ▼ | ▼ | [92] | |||||||||||||||||||||||
| DSC2 | p.R132C | △ | △ | ▼ | ▲ | ■ | ■ | ■ | ▼ | ▲ | ▲ | [88,185,186] | ||||||||||||||||||
| Heterozygous mutation (not shaded) | ▼ decreased relative to control | |||||||||||||||||||||||||||||
| Compound heterozygous mutation (shaded red) | ■ no change relative to control | |||||||||||||||||||||||||||||
| Homozygous mutation (shaded blue) | ▲ increased relative to control | |||||||||||||||||||||||||||||
| ▲■▼ membrane protein (immunofluoresence) | ||||||||||||||||||||||||||||||
| * Artificially introduced mutation (not patient derived) | ▲■▼ total protein abundance (Western blot) | |||||||||||||||||||||||||||||
| † Assumed heterozygous, although not specified | △□▽ total mRNA abundance (qPCR/RNA-seq) | |||||||||||||||||||||||||||||
| Molecular Data | Calcium | Force, Stress, & Contraction | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Mutation | RYR2/RyR2 | CACNA1C/Cav1.5 | SLC8A1/NCX1 | ATP2A2/SERCA2a | Diastolic intra Ca2+ | Systolic intra Ca2+ | Intra Ca2+ AUC | Intra Ca2+ time-to-peak | Intra Ca2+ decay time | Ca2+ transient frequency | Ca2+ sparks | EAD or DAD events | Peak force | Time-to-peak force | Contraction rate | Tissue diastolic length | Tissue shortening | Diastolic stress | Systolic stress | Systolic | Twitch stress | Contraction velocity | Relaxation duration | Aberrant contraction | Contraction asynchrony | Refs. |
| PKP2 | p.G828LfsX835 | □ | △ | □ | ▽ | ■ | [28] | ||||||||||||||||||||
| p.D109AfsX119 * | ▼ | ▼ | ▼ | [85] | |||||||||||||||||||||||
| p.D109AfsX119 * | ▼ | ▼ | ▼ | ||||||||||||||||||||||||
| p.D410fsX425 | ▼ | [86] | |||||||||||||||||||||||||
| p.D410fsX412 * | ▼ | ||||||||||||||||||||||||||
| DSG2 | p.G638R † | ■ | ■ | ▲ | [89] | ||||||||||||||||||||||
| p.D787MfsX807 | ▼ | ▼ | [59] | ||||||||||||||||||||||||
| p.R119X | ▼ | [90] | |||||||||||||||||||||||||
| DSP | p.R451G | ■ | ■ | ▲ | [91] | ||||||||||||||||||||||
| c.273+5G>A + p.R2229X2261 | ▼ | ▼ | ▲ | ▼ | [93] | ||||||||||||||||||||||
| DSC2 | p.R132C | ▼ | ▼ | ▼ | ▲ | ▲ | ▲ | ▲ | ■ | ▼ | ▲ | ▲ | [88,185,186] | ||||||||||||||
| Heterozygous mutation (not shaded) | ▼ decreased relative to control | ||||||||||||||||||||||||||
| Compound heterozygous mutation (shaded red) | ■ no change relative to control | ||||||||||||||||||||||||||
| Homozygous mutation (shaded blue) | ▲ increased relative to control | ||||||||||||||||||||||||||
| ▲■▼ membrane protein (immunofluoresence) | |||||||||||||||||||||||||||
| * Artificially introduced mutation (not patient derived) | ▲■▼ total protein abundance (Western blot) | ||||||||||||||||||||||||||
| † Assumed heterozygous, although not specified | △□▽ total mRNA abundance (qPCR/RNA-seq) | ||||||||||||||||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chua, C.J.; Morrissette-McAlmon, J.; Tung, L.; Boheler, K.R. Understanding Arrhythmogenic Cardiomyopathy: Advances through the Use of Human Pluripotent Stem Cell Models. Genes 2023, 14, 1864. https://doi.org/10.3390/genes14101864
Chua CJ, Morrissette-McAlmon J, Tung L, Boheler KR. Understanding Arrhythmogenic Cardiomyopathy: Advances through the Use of Human Pluripotent Stem Cell Models. Genes. 2023; 14(10):1864. https://doi.org/10.3390/genes14101864
Chicago/Turabian StyleChua, Christianne J., Justin Morrissette-McAlmon, Leslie Tung, and Kenneth R. Boheler. 2023. "Understanding Arrhythmogenic Cardiomyopathy: Advances through the Use of Human Pluripotent Stem Cell Models" Genes 14, no. 10: 1864. https://doi.org/10.3390/genes14101864
APA StyleChua, C. J., Morrissette-McAlmon, J., Tung, L., & Boheler, K. R. (2023). Understanding Arrhythmogenic Cardiomyopathy: Advances through the Use of Human Pluripotent Stem Cell Models. Genes, 14(10), 1864. https://doi.org/10.3390/genes14101864