
Genome-Wide Identification and Preliminary Functional Analysis of BAM (β-Amylase) Gene Family in Upland Cotton

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification and Phylogenetic Analysis of the GhBAM Gene Family in G. hirsutum
2.2. Chromosome Distribution, Synteny, Ka/Ks, and Phylogenetic Analysis of the GhBAM Family
2.3. Analysis of GhBAM Gene Domain and Conserved Motif
2.4. Subcellular Localization and Promoter Element Analysis of GhBAM7
2.5. GhBAM Gene Expression Analysis
3. Results
3.1. Identification and Sequence Retrieval of BAM Gene Family in Upland Cotton
3.2. BAM Family Sequence Comparison and Phylogenetic Tree Analysis
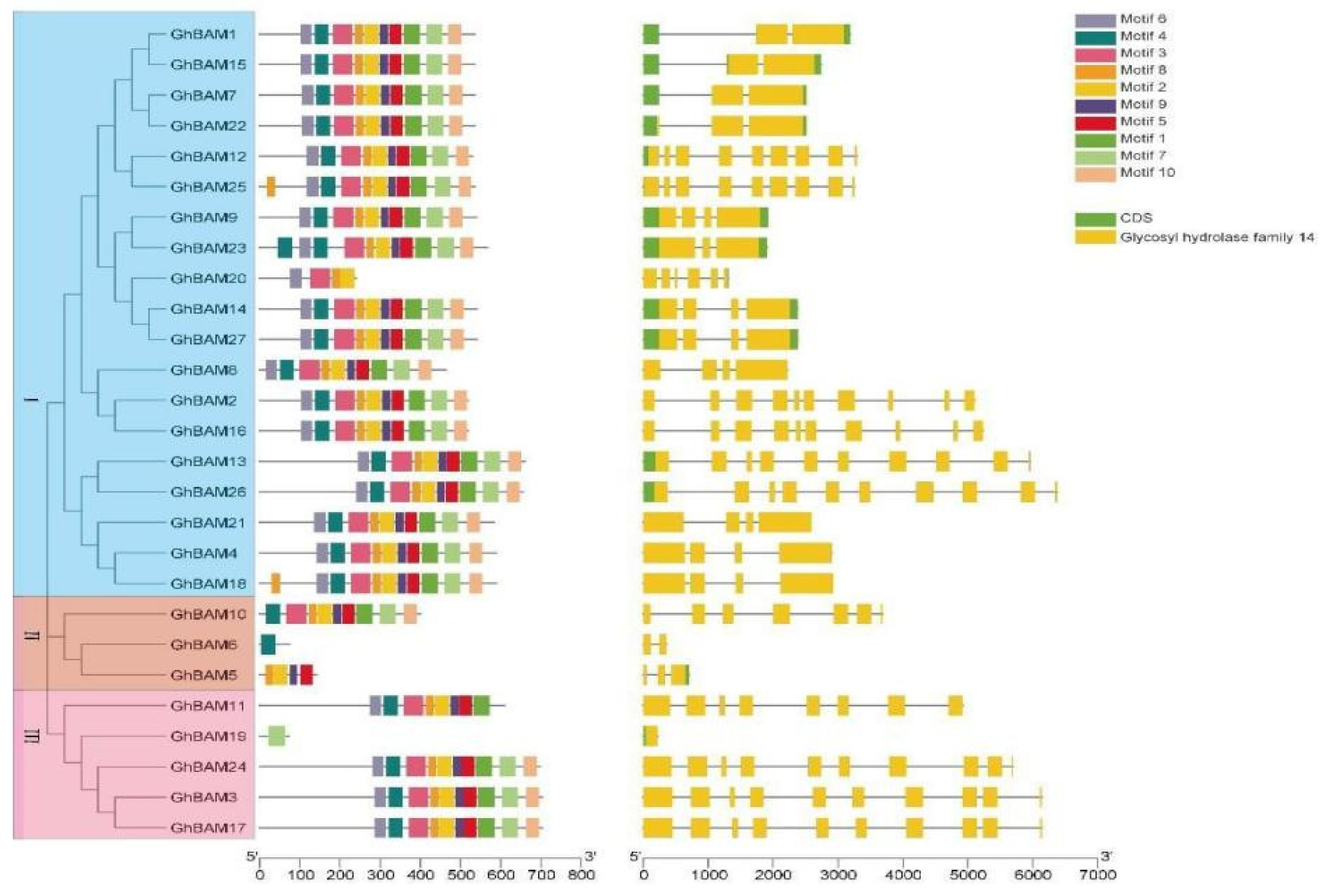
3.3. BAM Gene Structure and Conserved Motif Prediction
3.4. Prediction of Cis-Acting Elements in the GhBAM Gene Promoter Region
3.5. Chromosome Localization, Gene Replication, Collinearity Analysis, and Selection Pressure Analysis
3.6. Transcriptome Analysis
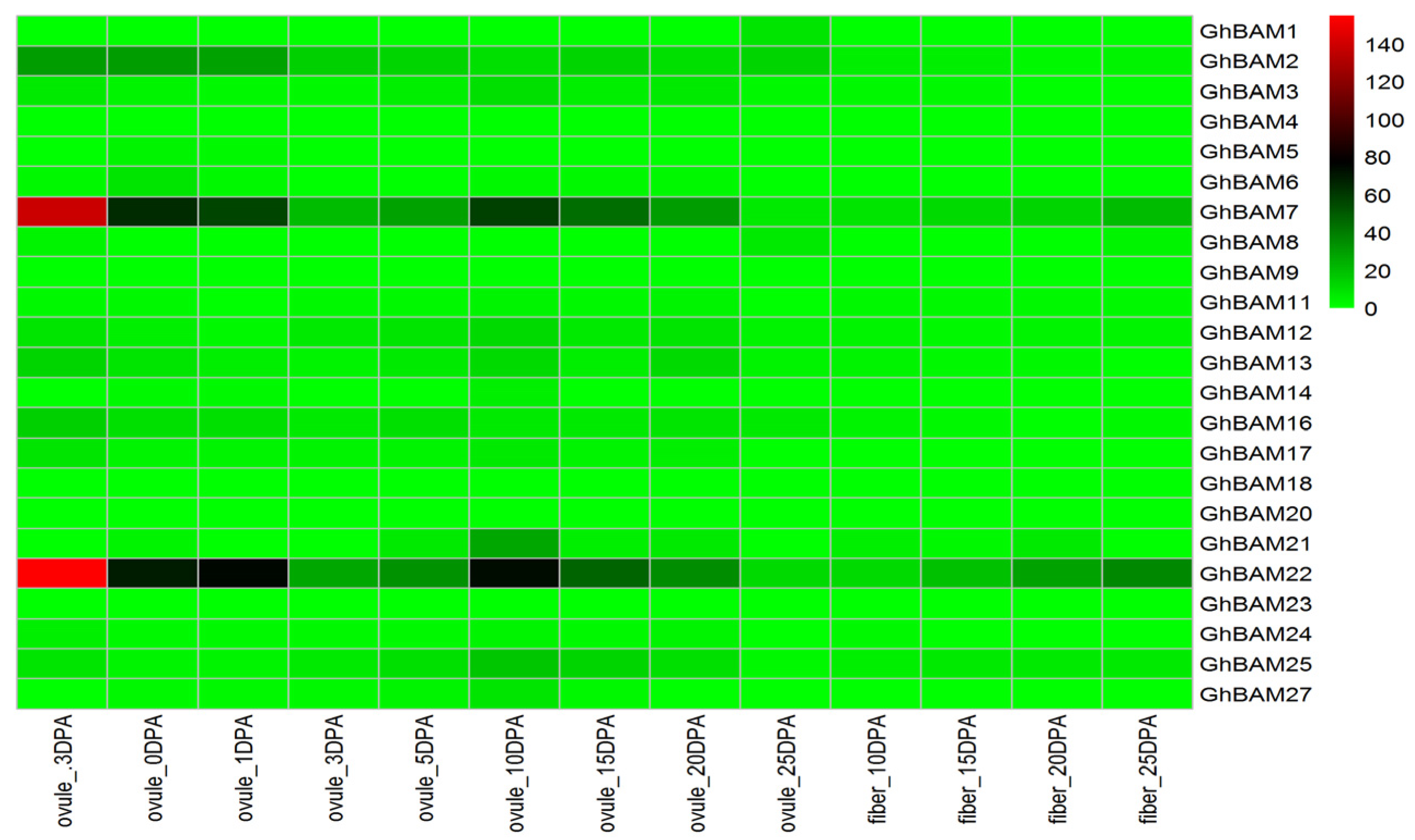
3.6.1. Analysis of GhBAM Gene Expression in Different Tissues of Upland Cotton
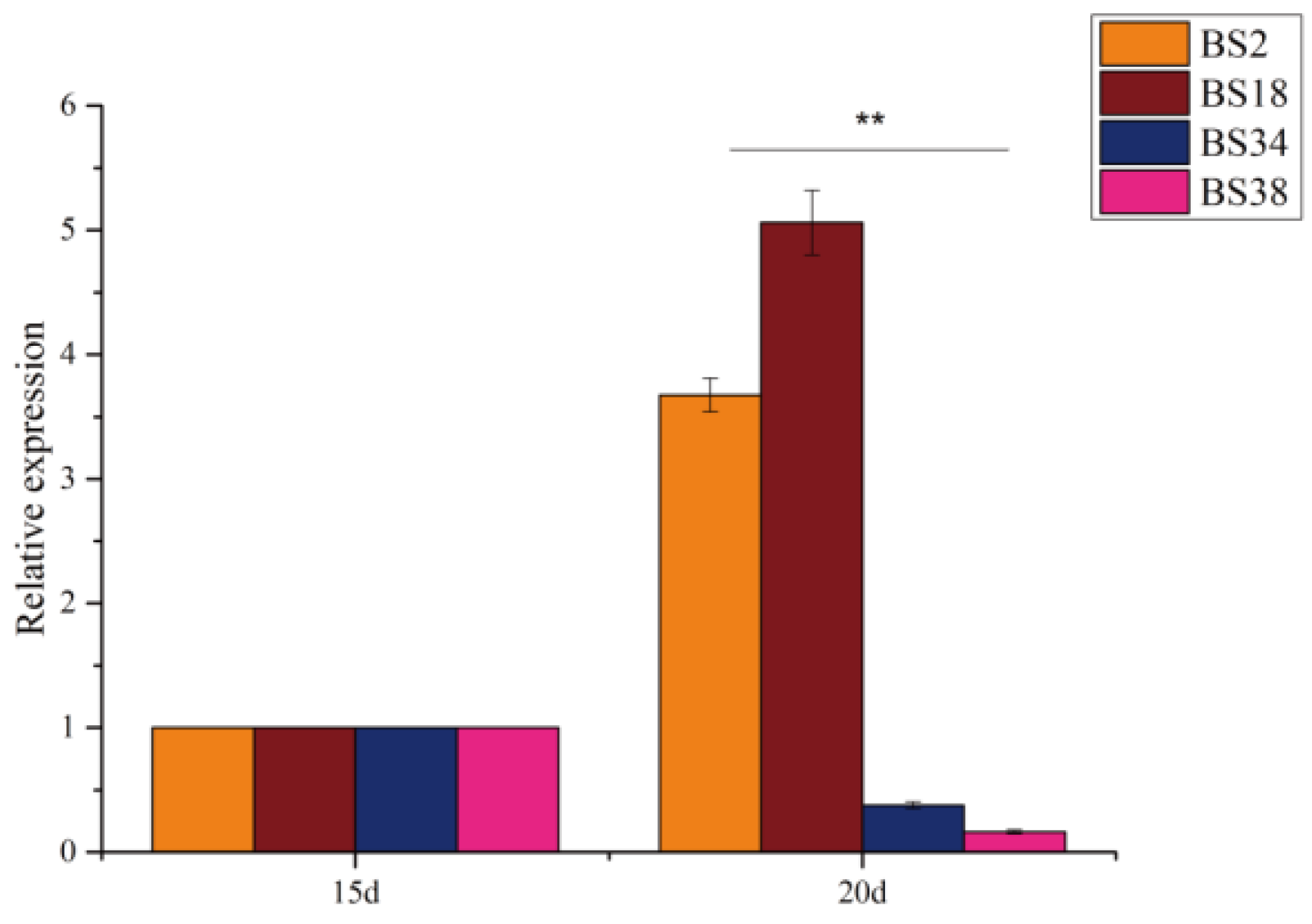
3.6.2. Transcriptional Expression Analysis of GhBAM Genes at 15 and 20 Days of Fiber Development in Extreme Materials
3.7. qRT-PCR Validation of the GhBAM Gene Family in Two Extreme Materials
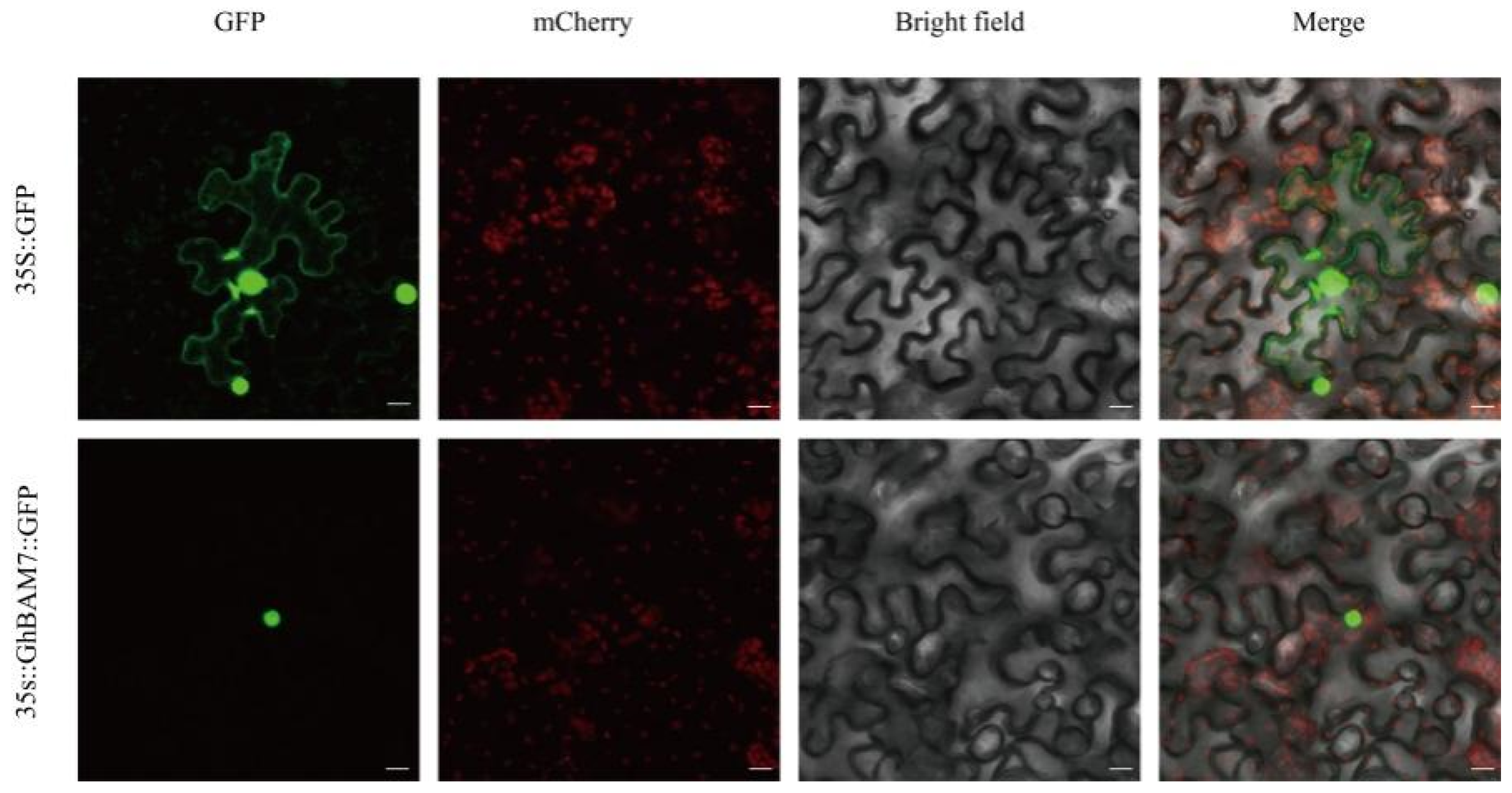
3.8. Subcellular Localization Analysis of GhBAM7 Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, D.; Wang, Y. β-amylase in developing apple fruits: Activities, amounts and subcellular localization. Sci. China Ser. C Life Sci. 2002, 45, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, M.; Santelia, D. Starch as a determinant of plant fitness under abiotic stress. New Phytol. 2017, 214, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Monroe, J.D. Involvement of five catalytically active Arabidopsis β-amylases in leaf starch metabolism and plant growth. Plant Direct 2020, 4, e00199. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Saini, J.S.; Mohan, A.; Brar, N.K.; Verma, S.; Sarao, N.K.; Gill, K.S. Comparative and evolutionary analysis of α-amylase gene across monocots and dicots. Funct. Integr. Genom. 2016, 16, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Zanella, M.; Borghi, G.L.; Pirone, C.; Thalmann, M.; Pazmino, D.; Costa, A.; Santelia, D.; Trost, P.; Sparla, F. β-amylase 1 (BAM1) degrades transitory starch to sustain proline biosynthesis during drought stress. J. Exp. Bot. 2016, 67, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Zeeman, S.C.; Smith, S.M. Starch degradation. Annu. Rev. Plant Biol. 2005, 56, 73–98. [Google Scholar] [CrossRef]
- Zeeman, S.C.; Delatte, T.; Messerli, G.; Umhang, M.; Stettler, M.; Mettler, T.; Streb, S.; Reinhold, H.; Kötting, O. Starch breakdown: Recent discoveries suggest distinct pathways and novel mechanisms. Funct. Plant Biol. 2007, 34, 465–473. [Google Scholar] [CrossRef]
- Valerio, C.; Costa, A.; Marri, L.; Issakidis-Bourguet, E.; Pupillo, P.; Trost, P.; Sparla, F. Thioredoxin-regulated β-amylase (BAM1) triggers diurnal starch degradation in guard cells, and in mesophyll cells under osmotic stress. J. Exp. Bot. 2011, 62, 545–555. [Google Scholar] [CrossRef]
- Zhao, L.Y.; Gong, X.; Gao, J.Z.; Dong, H.Z.; Zhang, S.L.; Tao, S.T.; Huang, X.S. Transcriptomic and evolutionary analyses of white pear (Pyrus bretschneideri) β-amylase genes reveals their importance for cold and drought stress responses. Gene 2019, 689, 102–113. [Google Scholar] [CrossRef]
- Fulton, D.C.; Stettler, M.; Mettler, T.; Vaughan, C.K.; Li, J.; Francisco, P.; Gil, M.; Reinhold, H.; Eicke, S.; Messerli, G.; et al. β-AMYLASE4, a noncatalytic protein required for starch breakdown, acts upstream of three active β-amylases in Arabidopsis chloroplasts. Plant Cell 2008, 20, 1040–1058. [Google Scholar] [CrossRef]
- Miao, H.X.; Sun, P.G.; Miao, Y.L.; Liu, J.H.; Zhang, J.B.; Jia, C.H.; Wang, J.Y.; Wang, Z.; Jin, Z.Q.; Xu, B.Y. Genome-wide identification and expression analysis of the β-amylase genes strongly associated with fruit development, ripening, and abiotic stress response in two banana cultivars. Front. Agric. Sci. Eng. 2016, 3, 346–356. [Google Scholar] [CrossRef]
- Jiang, S.Z.; Lian, H.; Xiong, Y.F.; Luo, K.J.; Su, X.Q.; Chen, S.P. Genome-wide identification and expression analysis of the β-amylase gene family in Castanea henry. J. For. Environ. 2021, 41, 545–553. [Google Scholar]
- Stitt, M.; Zeeman, S.C. Starch turnover: Pathways, regulation and role in growth. Curr. Opin. Plant Biol. 2012, 15, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Streb, S.; Zeeman, S.C. Starch metabolism in Arabidopsis. Arab. Book/Am. Soc. Plant Biol. 2012, 10, 73–98. [Google Scholar] [CrossRef]
- David, L.C.; Lee, S.K.; Bruderer, E.; Abt, M.R.; Fischer-Stettler, M.; Tschopp, M.A.; Solhaug, E.M.; Sanchez, K.; Zeeman, S.C. BETA-AMYLASE9 is a plastidial nonenzymatic regulator of leaf starch degradation. Plant Physiol. 2022, 188, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Weise, S.E.; Kim, K.S.; Stewart, R.P.; Sharkey, T.D. β-Maltose is the metabolically active anomer of maltose during transitory starch degradation. Plant Physiol. 2005, 137, 756–761. [Google Scholar] [CrossRef]
- Ziegler, P. Cereal Beta-Amylases. J. Cereal Sci. 1999, 29, 195–204. [Google Scholar] [CrossRef]
- Weise, S.E.; Weber, A.P.M.; Sharkey, T.D. Maltose is the major form of carbon exported from the chloroplast at night. Planta 2004, 218, 474–482. [Google Scholar] [CrossRef]
- Srivastava, G.; Kayastha, A.M. β-amylase from starchless seeds of Trigonella foenum-graecum and its local ization in germinating seeds. PLoS ONE 2014, 9, e88697. [Google Scholar] [CrossRef]
- Smith, S.M.; Fulton, D.C.; Chia, T.; Thorneycroft, D.; Chapple, A.; Dunstan, H.; Hylton, C.; Zeeman, S.C.; Smith, A.M. Diurnal Changes in the Transcriptome Encoding Enzymes of Starch Metabolism Provide Evidence for Both Transcriptional and Posttranscriptional Regulation of Starch metabolism in Arabidopsis leaves. Plant Physiol. 2004, 136, 2687–2699. [Google Scholar] [CrossRef]
- Koide, T.; Ohnishi, Y.; Horinouchi, S. Characterization of recombinant β-amylases from Oryza sativa. Biosci. Biotechnol. Biochem. 2011, 75, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Mason-Gamer, R.J. The β-amylase genes of grasses and a phylogenetic analysis of the Triticeae (Poaceae). Am. J. Bot. 2005, 92, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhang, H.L.; Liu, J.; Reid, S.; Liu, T.F.; Xu, S.J.; Tian, Z.D.; Sonnewald, U.; Song, B.T.; Xie, C.H. Amylases St Amy23, St BAM1 and St BAM9 regulate cold-induced sweetening of potato tubers in distinct ways. J. Exp. Bot. 2017, 68, 2317–2331. [Google Scholar] [CrossRef]
- Niittyla, T.; Messerli, G.; Trevisan, M.; Chen, J.; Smith, A.M.; Zeeman, S.C. A previously unknown maltose transporter essential for starch degradation in leaves. Science 2004, 303, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, M.; Coiro, M.; Meier, T.; Wicker, T.; Zeeman, S.C.; Santelia, D. The evolution of functional complexity within the β-amylase gene family in land plants. BMC Evol. Biol. 2019, 19, 66. [Google Scholar] [CrossRef]
- Monroe, J.D.; Breault, J.S.; Pope, L.E.; Torres, C.E.; Gebrejesus, T.B.; Berndsen, C.E.; Storm, A.R. Arabidopsis β-amylase2 is a K+-requiring, catalytic tetramer with sigmoidal kinetics. Plant Physiol. 2017, 175, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Monroe, J.D.; Storm, A.R. The Arabidopsis β-amylase (BAM) gene family: Diversity of form and function. Plant Sci. 2018, 276, 163–170. [Google Scholar] [CrossRef]
- Ma, Z.Y.; He, S.P.; Wang, X.F.; Sun, J.L.; Zhang, Y.; Zhang, G.Y.; Wu, L.Q.; Li, Z.K.; Liu, Z.H.; Sun, G.F.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef]
- Shan, C.M.; Shangguan, X.X.; Zhao, B.; Zhang, X.F.; Chao, L.M.; Yang, C.Q.; Wang, L.J.; Zhu, H.Y.; Zeng, Y.D.; Guo, W.Z.; et al. Control of cotton fibre elongation by a homeodomain transcription factor Gh HOX3. Nat. Commun. 2014, 5, 5519. [Google Scholar] [CrossRef]
- Tang, W.X.; Tu, L.L.; Yang, X.Y.; Tan, J.F.; Deng, F.L.; Hao, J.; Guo, K.; Lindsey, K.; Zhang, X.L. The calcium sensor Gh CaM7 promotes cotton fiber elongation by modulating reactive oxygen species (ROS) production. New Phytol. 2014, 202, 509–520. [Google Scholar] [CrossRef]
- Hajihashemi, S.; Skalicky, M.; Brestic, M.; Pavla, V. Cross-talk between nitric oxide, hydrogen peroxide and calcium in salt-stressed Chenopodium quinoa Willd. at seed germination stage. Plant Physiol. Biochem. 2020, 154, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Vinje, M.A.; Willis, D.K.; Duke, S.H.; Henson, C.A. Differential expression of two b-amylase genes (Bmy1 and Bmy2) in developing and mature barley grain. Planta 2011, 233, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.H.; Chen, J.D.; Liu, B.L.; Zhang, Z.Y.; Guan, X.Y.; Chen, S.Q.; Zhou, B.L.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.P.; Garvin, D.F.; Mockler, T.C.; Schmutz, J.; Rokhsar, D.; Bevan, M.W.; Lail, K. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar]
- Vatansever, R.; Koc, I.; Ozyigit, I.I.; Sen, U.; Uras, M.E.; Anjum, N.A.; Pereira, E.; Filiz, E. Genome-wide identification and expression analysis of sulfate transporter (SULTR) genes in potato (Solanum tuberosum L.). Planta 2016, 244, 1167–1183. [Google Scholar] [CrossRef]
- Li, J.; Francisco, P.; Zhou, W.X.; Edner, C.; Steup, M.; Ritte, G.; Bond, C.S.; Smith, S.M. Catalytically-inactive β-amylase BAM4 required for starch breakdown in Arabidopsis leaves is a starch-binding-protein. Arch. Biochem. Biophys. 2009, 489, 92–98. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Sun, F.; Wang, P.; Yusuyin, M.; Kuerban, W.; Lai, C.; Li, C.; Ma, J.; Xiao, F. Genome-Wide Identification and Preliminary Functional Analysis of BAM (β-Amylase) Gene Family in Upland Cotton. Genes 2023, 14, 2077. https://doi.org/10.3390/genes14112077
Yang Y, Sun F, Wang P, Yusuyin M, Kuerban W, Lai C, Li C, Ma J, Xiao F. Genome-Wide Identification and Preliminary Functional Analysis of BAM (β-Amylase) Gene Family in Upland Cotton. Genes. 2023; 14(11):2077. https://doi.org/10.3390/genes14112077
Chicago/Turabian StyleYang, Yanlong, Fenglei Sun, Penglong Wang, Mayila Yusuyin, Wumaierjiang Kuerban, Chengxia Lai, Chunping Li, Jun Ma, and Fei Xiao. 2023. "Genome-Wide Identification and Preliminary Functional Analysis of BAM (β-Amylase) Gene Family in Upland Cotton" Genes 14, no. 11: 2077. https://doi.org/10.3390/genes14112077
APA StyleYang, Y., Sun, F., Wang, P., Yusuyin, M., Kuerban, W., Lai, C., Li, C., Ma, J., & Xiao, F. (2023). Genome-Wide Identification and Preliminary Functional Analysis of BAM (β-Amylase) Gene Family in Upland Cotton. Genes, 14(11), 2077. https://doi.org/10.3390/genes14112077