


Prognostic Value of Genotype–Phenotype Correlations in X-Linked Myotubular Myopathy and the Use of the Face2Gene Application as an Effective Non-Invasive Diagnostic Tool

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Subjects and Methods
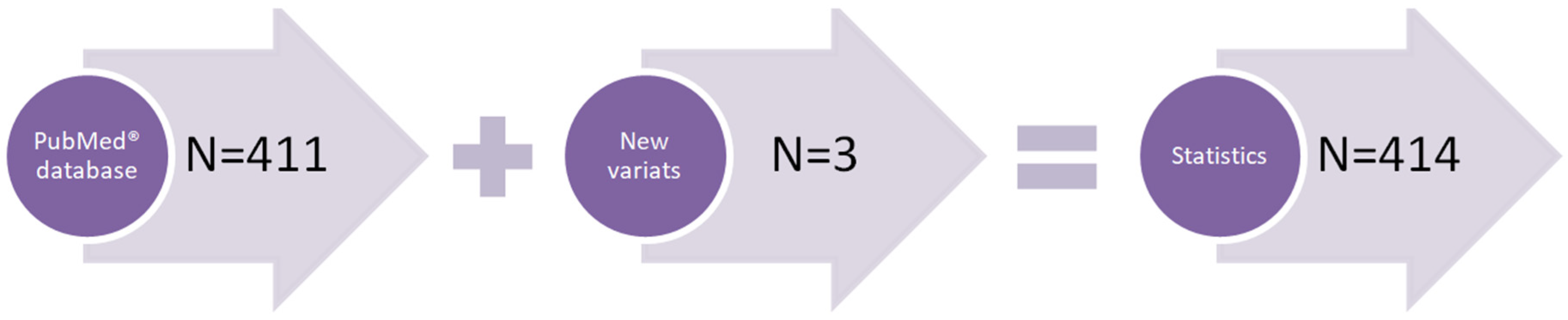
2.1. Subjects Involved in the Statistical Analysis
2.2. Phenotype Severity
2.3. Variant Evaluation
2.4. Facial Gestalt Analysis
2.5. Statistical Analysis of Genotype–Phenotype Correlations
3. Results
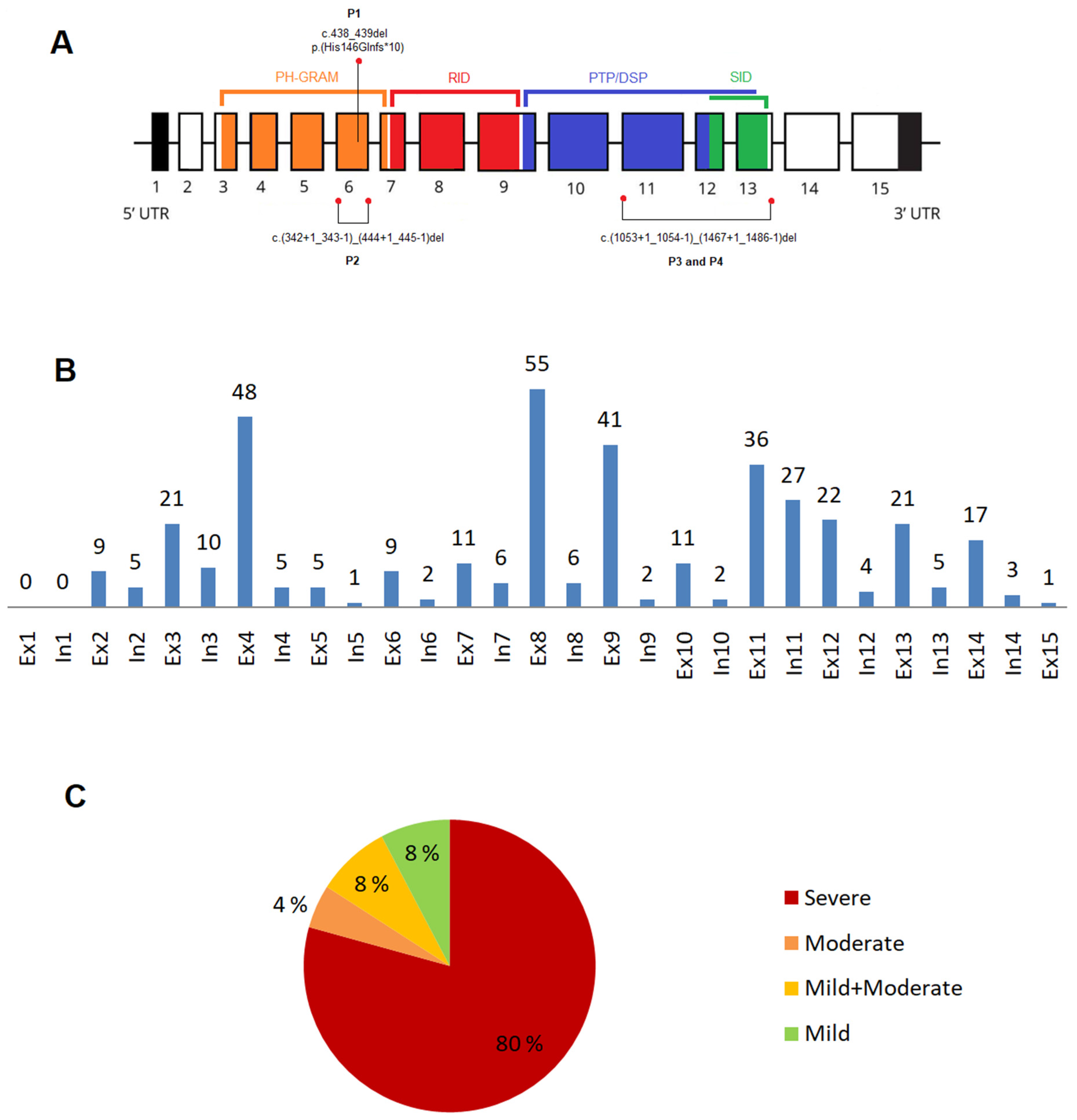
3.1. Novel Variants in the MTM1 Gene
3.2. XLMTM Cohort: Variants Type and Their Distribution
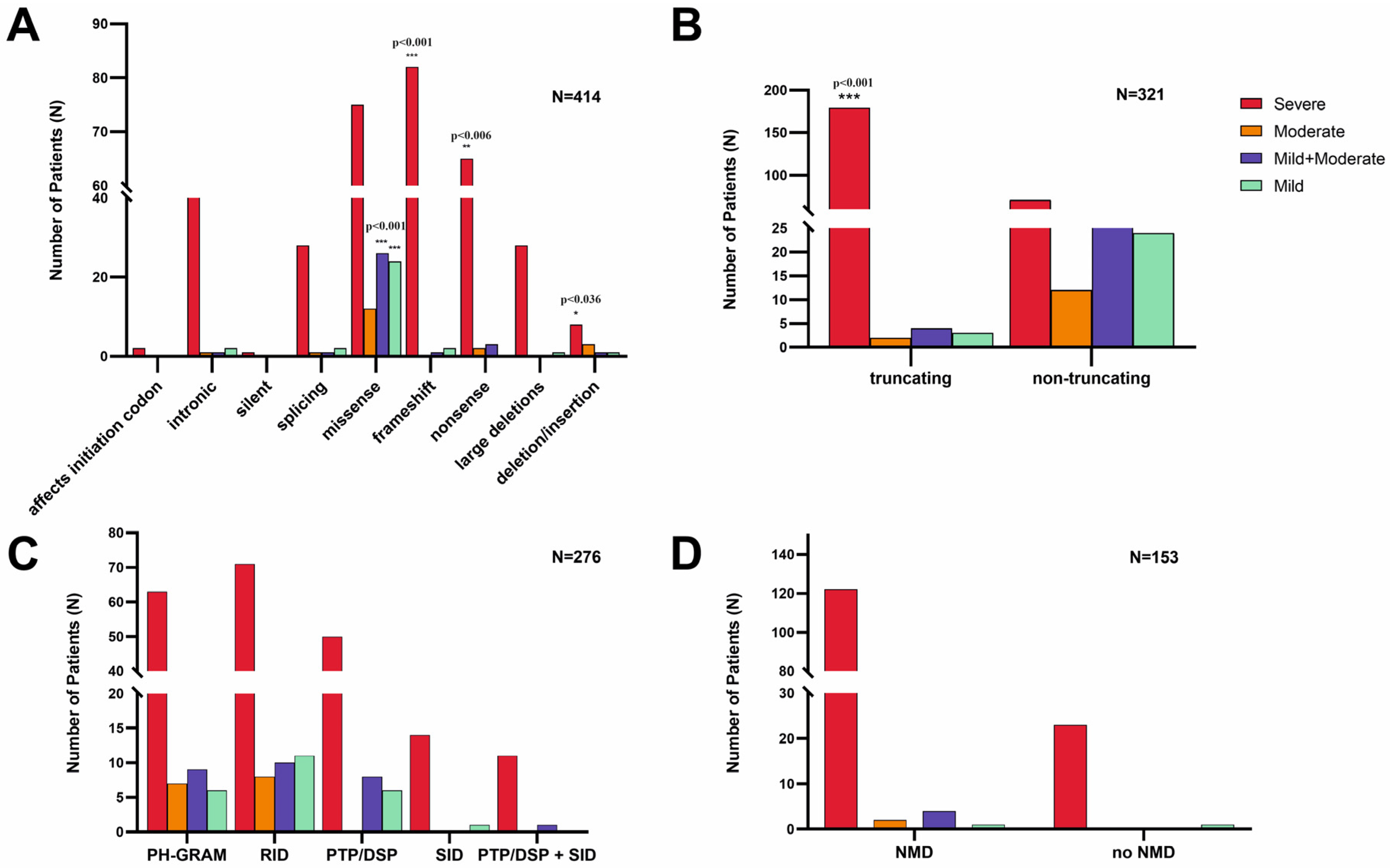
3.2.1. Exonic and Intronic Variants
3.2.2. Variants in the MTM1-Specific Functional Domain
3.2.3. Truncating Effect and Nonsense-Mediated mRNA Decay
3.2.4. Phenotype Severity and Variant Types
3.2.5. Recurrent Variants in the MTM1 Gene
3.2.6. Inter-Individual Phenotype Variability
3.3. Genotype–Phenotype Correlations
3.4. Facial Gestalt Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, G.S.; Maehama, T.; Dixon, J.E. Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc. Natl. Acad. Sci. USA 2000, 97, 8910–8915. [Google Scholar] [CrossRef] [PubMed]
- Robinson, F.L.; Dixon, J.E. Myotubularin phosphatases: Policing 3-phosphoinositides. Trends Cell Biol. 2006, 16, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Bertazzi, D.; De Craene, J.-O.; Friant, S. Myotubularin MTM1 Involved in Centronuclear Myopathy and its Roles in Human and Yeast Cells. J. Mol. Genet. Med. 2015, 8, 1–6. [Google Scholar] [CrossRef]
- Laporte, J.; Guiraud-Chaumeil, C.; Tanner, S.M.; Blondeau, F.; Hu, L.J.; Vicaire, S.; Liechti-Gallati, S.; Mandel, J.L. Genomic organization of the MTM1 gene implicated in X-linked myotubular myopathy. Eur. J. Hum. Genet. 1998, 6, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Spiro, A.J.; Shy, G.M.; Gonatas, N.K. Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Arch. Neurol. 1966, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Laporte, J.; Blondeau, F.; Buj-Bello, A.; Mandel, J.L. The myotubularin family: From genetic disease to phosphoinositide metabolism. Trends Genet. 2001, 17, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Herman, G.E.; Finegold, M.; Zhao, W.; de Gouyon, B.; Metzenberg, A. Medical complications in long-term survivors with X-linked myotubular myopathy. J. Pediatr. 1999, 134, 206–214. [Google Scholar] [CrossRef]
- McEntagart, M.; Parsons, G.; Buj-Bello, A.; Biancalana, V.; Fenton, I.; Little, M.; Krawczak, M.; Thomas, N.; Herman, G.; Clarke, A.; et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul. Disord. 2002, 12, 939–946. [Google Scholar] [CrossRef]
- Oliveira, J.; Oliveira, M.E.; Kress, W.; Taipa, R.; Pires, M.M.; Hilbert, P.; Baxter, P.; Santos, M.; Buermans, H.; den Dunnen, J.T.; et al. Expanding the MTM1 mutational spectrum: Novel variants including the first multi-exonic duplication and development of a locus-specific database. Eur. J. Hum. Genet. 2013, 21, 540–549. [Google Scholar] [CrossRef]
- Dowling, J.J.; Lawlor, M.W.; Das, S. X-Linked Myotubular Myopathy. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2018; 25 February 2002; [Updated 23 August 2018]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1432/ (accessed on 1 November 2023).
- Lawlor, M.W.; Dowling, J.J. X-linked myotubular myopathy. Neuromuscul. Disord. 2021, 31, 1004–1012. [Google Scholar] [CrossRef]
- Motoki, T.; Fukuda, M.; Nakano, T.; Matsukage, S.; Fukui, A.; Akiyoshi, S.; Hayashi, Y.K.; Ishii, E.; Nishino, I. Fatal hepatic hemorrhage by peliosis hepatis in X-linked myotubular myopathy: A case report. Neuromuscul. Disord. 2013, 23, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Amburgey, K.; Tsuchiya, E.; de Chastonay, S.; Glueck, M.; Alverez, R.; Nguyen, C.T.; Rutkowski, A.; Hornyak, J.; Beggs, A.H.; Dowling, J.J. A natural history study of X-linked myotubular myopathy. Neurology 2017, 89, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Beggs, A.H.; Byrne, B.J.; De Chastonay, S.; Haselkorn, T.; Hughes, I.; James, E.S.; Kuntz, N.L.; Simon, J.; Swanson, L.C.; Yang, M.L.; et al. A multicenter, retrospective medical record review of X-linked myotubular myopathy: The recensus study. Muscle Nerve 2018, 57, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Gurovich, Y.; Hanani, Y.; Bar, O. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 2019, 25, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Laporte, J.; Biancalana, V.; Tanner, S.M.; Kress, W.; Schneider, V.; Wallgren-Pettersson, C.; Herger, F.; Buj-Bello, A.; Blondeau, F.; Liechti-Gallati, S.; et al. MTM1 mutations in X-linked myotubular myopathy. Hum. Mutat. 2000, 15, 393–409. [Google Scholar] [CrossRef]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Annoussamy, M.; Lilien, C.; Gidaro, T.; Gargaun, E.; Chê, V.; Schara, U.; Gangfuß, A.; D’Amico, A.; Dowling, J.J.; Darras, B.T.; et al. X-linked myotubular myopathy: A prospective international natural history study. Neurology 2019, 92, e1852–e1867. [Google Scholar] [CrossRef]
- Fattori, F.; Maggi, L.; Bruno, C.; Cassandrini, D.; Codemo, V.; Catteruccia, M.; Tasca, G.; Berardinelli, A.; Magri, F.; Pane, M.; et al. Centronuclear myopathies: Genotype-phenotype correlation and frequency of defined genetic forms in an Italian cohort. J. Neurol. 2015, 262, 1728–1740. [Google Scholar] [CrossRef]
- Biancalana, V.; Scheidecker, S.; Miguet, M.; Laquerrière, A.; Romero, N.B.; Stojkovic, T.; Abath Neto, O.; Mercier, S.; Voermans, N.; Tanner, L.; et al. Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: Delineating diagnostic clues. Acta Neuropathol. 2017, 134, 889–904. [Google Scholar] [CrossRef]
- Tsai, T.C.; Horinouchi, H.; Noguchi, S.; Minami, N.; Murayama, K.; Hayashi, Y.K.; Nonaka, I.; Nishino, I. Characterization of MTM1 mutations in 31 Japanese families with myotubular myopathy, including a patient carrying 240 kb deletion in Xq28 without male hypogenitalism. Neuromuscul. Disord. 2005, 15, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, V.; Caron, O.; Gallati, S.; Baas, F.; Kress, W.; Novelli, G.; D’Apice, M.R.; Lagier-Tourenne, C.; Buj-Bello, A.; Romero, N.B.; et al. Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum. Genet. 2003, 112, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Herman, G.E.; Kopacz, K.; Zhao, W.; Mills, P.L.; Metzenberg, A.; Das, S. Characterization of mutations in fifty North American patients with X-linked myotubular myopathy. Hum. Mutat. 2002, 19, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Laporte, J.; Guiraud-Chaumeil, C.; Vincent, M.C.; Mandel, J.L.; Tanner, S.M.; Liechti-Gallati, S.; Wallgren-Pettersson, C.; Dahl, N.; Kress, W.; Bolhuis, P.A.; et al. Mutations in the MTM1 gene implicated in X-linked myotubular myopathy. ENMC International Consortium on Myotubular Myopathy. European Neuro-Muscular Center. Hum. Mol. Genet. 1997, 6, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Buj-Bello, A.; Biancalana, V.; Moutou, C.; Laporte, J.; Mandel, J.L. Identification of novel mutations in the MTM1 gene causing severe and mild forms of X-linked myotubular myopathy. Hum. Mutat. 1999, 14, 320–325. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Bar-Haim, A.; Moosa, S.; Ehmke, N.; Gripp, K.W.; Pantel, J.T.; Danyel, M.; Mensah, M.A.; Horn, D.; Rosnev, S.; et al. GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat. Genet. 2022, 54, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Liechti-Gallati, S.; Müller, B.; Grimm, T.; Kress, W.; Müller, C.; Boltshauser, E.; Moser, H.; Braga, S. X-linked centronuclear myopathy: Mapping the gene to Xq28. Neuromuscul. Disord. 1991, 1, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Tanner, S.M.; Schneider, V.; Thomas, N.S.; Clarke, A.; Lazarou, L.; Liechti-Gallati, S. Characterization of 34 novel and six known MTM1 gene mutations in 47 unrelated X-linked myotubular myopathy patients. Neuromuscul. Disord. 1999, 9, 41–49. [Google Scholar] [CrossRef]
- de Gouyon, B.M.; Zhao, W.; Laporte, J.; Mandel, J.L.; Metzenberg, A.; Herman, G.E. Characterization of mutations in the myotubularin gene in twenty six patients with X-linked myotubular myopathy. Hum. Mol. Genet. 1997, 6, 1499–1504. [Google Scholar] [CrossRef]
- Al-Hashim, A.; Gonorazky, H.D.; Amburgey, K.; Das, S.; Dowling, J.J. A novel intronic mutation in MTM1 detected by RNA analysis in a case of X-linked myotubular myopathy. Neurol. Genet. 2017, 3, e182. [Google Scholar] [CrossRef]
- Savarese, M.; Musumeci, O.; Giugliano, T.; Rubegni, A.; Fiorillo, C.; Fattori, F.; Torella, A.; Battini, R.; Rodolico, C.; Pugliese, A.; et al. Novel findings associated with MTM1 suggest a higher number of female symptomatic carriers. Neuromuscul. Disord. 2016, 26, 292–299. [Google Scholar] [CrossRef]
- Barth, P.G.; Dubowitz, V. X-linked myotubular myopathy. A long-term follow-up study. Eur. J. Paediatr. Neurol. 1998, 2, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hoffjan, S.; Thiels, C.; Vorgerd, M.; Neuen-Jacob, E.; Epplen, J.T.; Kress, W. Extreme phenotypic variability in a German family with X-linked myotubular myopathy associated with E404K mutation in MTM1. Neuromuscul. Disord. 2006, 16, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Bertazzi, D.L.; Tronchère, H.; Hnia, K.; Chicanne, G.; Rinaldi, B.; Cowling, B.S.; Ferry, A.; Klaholz, B.; Payrastre, B.; et al. Phosphatase-dead myotubularin ameliorates X-linked centronuclear myopathy phenotypes in mice. PLoS Genet. 2012, 8, e1002965. [Google Scholar] [CrossRef]
- Pierson, C.R.; Agrawal, P.B.; Blasko, J.; Beggs, A.H. Myofiber size correlates with MTM1 mutation type and outcome in X-linked myotubular myopathy. Neuromuscul. Disord. 2007, 17, 562–568. [Google Scholar] [CrossRef]
- Bryen, S.J.; Oates, E.C.; Evesson, F.J.; Lu, J.K.; Waddell, L.B.; Joshi, H.; Ryan, M.M.; Cummings, B.B.; McLean, C.A.; MacArthur, D.G.; et al. Pathogenic deep intronic MTM1 variant activates a pseudo-exon encoding a nonsense codon resulting in severe X-linked myotubular myopathy. Eur. J. Hum. Genet. 2021, 29, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Paila, U.; Chapman, B.A.; Kirchner, R.; Quinlan, A.R. GEMINI: Integrative exploration of genetic variation and genome annotations. PLoS Comput. Biol. 2013, 9, e1003153. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Rivier, F.; Hamroun, D. The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2017, 27, 1152–1183. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef]
- den Dunnen, J.T.; Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum. Mutat. 2000, 15, 7–12. [Google Scholar] [CrossRef]
- Dubowitz, V.; Sewry, C.A.; Oldfors, A. Muscle Biopsy. A Practical Approach, 5th ed.; SAUNDERS Elsevier: Amsterdam, The Netherlands, 2020; Printed in China; pp. 1–660. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Ancestry (Ethnicity) | MTM1 Variant | Type | Exon | Domain | ACMG Classification | Origin | Phenotype |
|---|---|---|---|---|---|---|---|---|
| P1/F1 M | Slovakia (Caucasian) | c.438_439delCA (p.His146Glnfs*10) | Frameshift | ex6 | PH-GRAM | Pathogenic | Maternal | Severe |
| P2/F2 M | Austria (Caucasian) | c.(342+1_343-1)_(444+1_445-1)del p.(Asp115_Leu148del) | In-frame deletion | ex6 | PH-GRAM | Likely pathogenic | Maternal | Severe |
| P3/F3 M | Austria (Caucasian) | c.(1053+1_1054-1)_(1467+1_1468-1)del; p.(Leu352_Gln489)del | In-frame deletion | ex11-13 | PTP/DSP-SID | Likely pathogenic | Maternal | Severe |
| P4/F3 M | Austria (Caucasian) | c.(1053+1_1054-1)_(1467+1_1468-1)del; p.(Leu352_Gln489)del | In-frame deletion | ex11-13 | PTP/DSP-SID | Likely pathogenic | Maternal | Severe |
| Variant | Type | Exon/intron | Domain | Count | Phenotype | Reference |
|---|---|---|---|---|---|---|
| c.98_103del, p.(Glu33_Ala34del) | Deletion | ex3 | PH-GRAM | 2 | 1S/1Mi | [19,20] |
| c.109C>T, p.(Arg37 *) | Nonsense | ex3 | PH-GRAM | 9 | 8S/1Mo | [21,22,23,24,25,26] |
| c.139_141del, p.(Lys47del) | Deletion | ex4 | PH-GRAM | 5 | 4S/1Mo | [23,25,27,28] |
| c.142G>T, p.(Glu48 *) | Nonsense | ex4 | PH-GRAM | 2 | 1S/1Mo | [7,29] |
| c.205C>T, p.(Arg69Cys) | Missense | ex4 | PH-GRAM | 12 | 2S/1Mo/6M/3Mi | [16,19,20,24,30] |
| c.232-26_232-23del | Intronic | in4 | - | 2 | 1S/1Mo | [19,31] |
| c.590C>T, p.(Thr197Ile) | Missense | ex8 | RID | 3 | 1S/1Mo/1Mi | [16,20] |
| c.614C>T, p.(Pro205Leu) | Missense | ex8 | RID | 9 | 8S/1Mi | [19,22,23,24,29] |
| c.679G>A, p.(Val227Met) | Missense | ex9 | RID | 3 | 1S/1M/1Mi | [16,19,24] |
| c.695A>G, p.(His232Arg) | Missense | ex9 | RID | 2 | 1S/1Mo | [19,29] |
| c.721C>T, p.(Arg241Cys) | Missense | ex9 | RID | 13 | 3S/1Mo/5M/4Mi | [16,19,25,26,28,29] |
| c.1262G>A, p.(Arg421Gln) | Missense | ex12 | PTP/DSP | 8 | 7S/1M | [16,23,24,25,30] |
| c.1558C>T, p.(Arg520 *) | Nonsense | ex14 | - | 3 | 1S/2M | [16,18] |
| Binary Comparison | No. of Cases | Mean AUC | AUC SD | p Value for AUC |
|---|---|---|---|---|
| XLMTM vs. Unaffected | 14 vs. 11 | 1.00 | 0.01 | 0.001 |
| XLMTM vs. MD1 | 14 vs. 10 | 0.88 | 0.07 | 0.074 |
| XLMTM vs. NMDs | 14 vs. 11 | 0.63 | 0.09 | 0.229 |
| Unaffected vs. NMDs | 11 vs. 11 | 0.99 | 0.02 | 0.023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kušíková, K.; Šoltýsová, A.; Ficek, A.; Feichtinger, R.G.; Mayr, J.A.; Škopková, M.; Gašperíková, D.; Kolníková, M.; Ornig, K.; Kalev, O.; et al. Prognostic Value of Genotype–Phenotype Correlations in X-Linked Myotubular Myopathy and the Use of the Face2Gene Application as an Effective Non-Invasive Diagnostic Tool. Genes 2023, 14, 2174. https://doi.org/10.3390/genes14122174
Kušíková K, Šoltýsová A, Ficek A, Feichtinger RG, Mayr JA, Škopková M, Gašperíková D, Kolníková M, Ornig K, Kalev O, et al. Prognostic Value of Genotype–Phenotype Correlations in X-Linked Myotubular Myopathy and the Use of the Face2Gene Application as an Effective Non-Invasive Diagnostic Tool. Genes. 2023; 14(12):2174. https://doi.org/10.3390/genes14122174
Chicago/Turabian StyleKušíková, Katarína, Andrea Šoltýsová, Andrej Ficek, René G. Feichtinger, Johannes A. Mayr, Martina Škopková, Daniela Gašperíková, Miriam Kolníková, Karoline Ornig, Ognian Kalev, and et al. 2023. "Prognostic Value of Genotype–Phenotype Correlations in X-Linked Myotubular Myopathy and the Use of the Face2Gene Application as an Effective Non-Invasive Diagnostic Tool" Genes 14, no. 12: 2174. https://doi.org/10.3390/genes14122174
APA StyleKušíková, K., Šoltýsová, A., Ficek, A., Feichtinger, R. G., Mayr, J. A., Škopková, M., Gašperíková, D., Kolníková, M., Ornig, K., Kalev, O., Weis, S., & Weis, D. (2023). Prognostic Value of Genotype–Phenotype Correlations in X-Linked Myotubular Myopathy and the Use of the Face2Gene Application as an Effective Non-Invasive Diagnostic Tool. Genes, 14(12), 2174. https://doi.org/10.3390/genes14122174