
Whole Genome Analysis of SNV and Indel Polymorphism in Common Marmosets (Callithrix jacchus)

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Marmoset Genome Assembly
2.2. DNA Sample Preparation for Studies of Within-Species Variation
2.3. Fibroblast Derivation from Ear Biopsies
2.4. Collection of Hair Follicles and Isolation of DNA from SNPRC Marmosets
2.5. WGS Library Preparation
2.6. Illumina Whole Genome Sequencing
2.7. Mapping and Variant Calling
2.8. PCA and ADMIXTURE Analyses
2.9. Genome Diversity
2.10. Variant Orthologs in Human and Pathogenicity Predictions
2.11. Marmoset SNVs Orthologous to ClinVar SNVs
2.12. Neurodevelopmental Gene Variants
2.13. Alzheimer’s Disease Gene Variants
2.14. Callitrichine Size and Reproduction-Related Genes
3. Results
3.1. Genome Assembly and Gene Annotation
3.2. Single Nucleotide Variants
3.3. Indel Variants
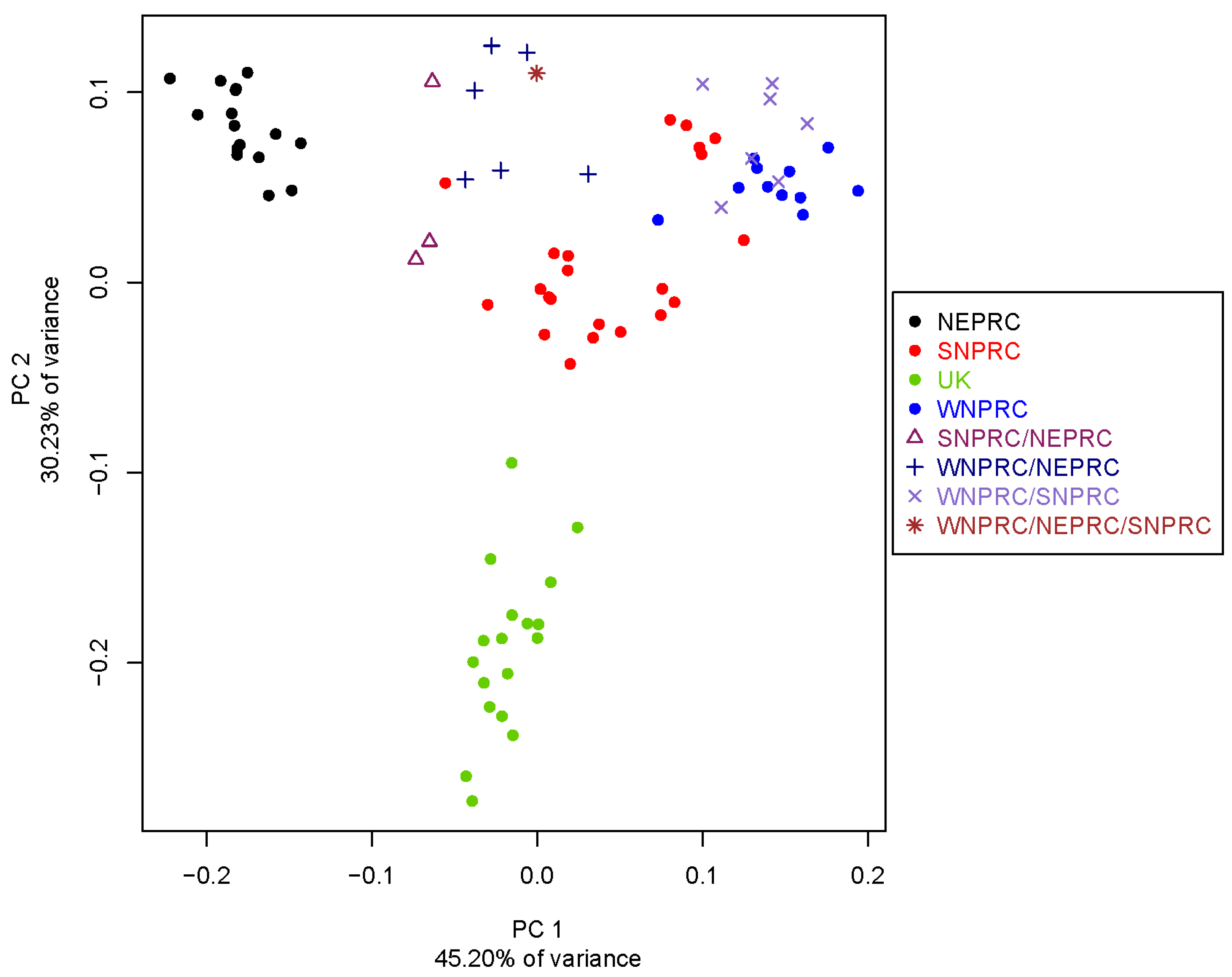
3.4. Population Genetics
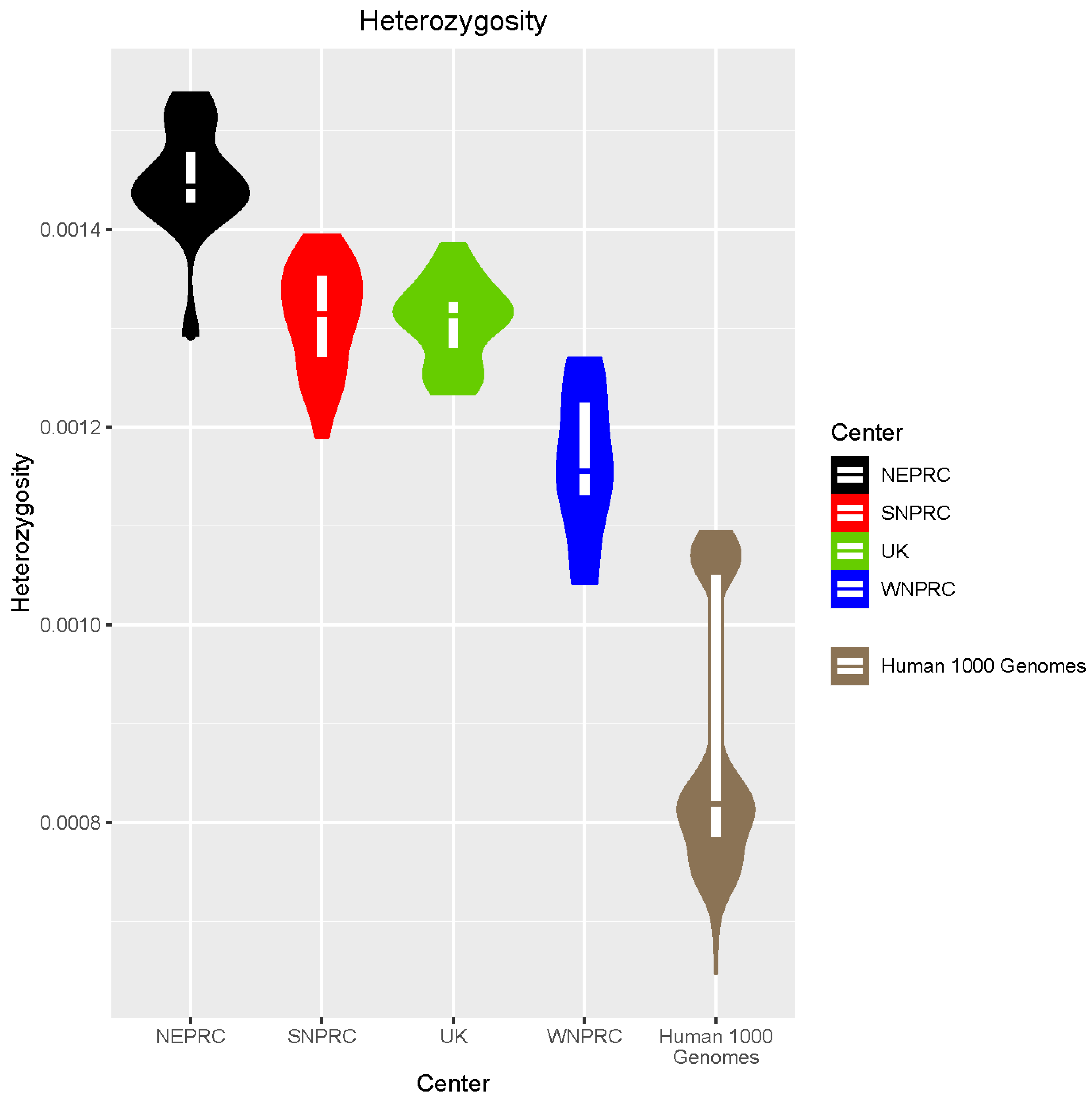
3.5. Genome Diversity
3.6. Marmoset SNVs Orthologous to ClinVar SNVs
3.7. Neurodevelopmental Gene Variants
3.8. Alzheimer’s Disease Gene Variants
3.9. Callitrichine Size and Reproduction-Related Genes
3.10. Marmoset Variants UCSC Genome Browser Track Hub
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tardif, S.D.; Power, M.L.; Ross, C.N.; Rutherford, J.N. Body mass growth in common marmosets: Toward a model of pediatric obesity. Am. J. Phys. Anthropol. 2013, 150, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Tardif, S.D.; Vinar, T.; Wildman, D.E.; Rutherford, J.N.; Rogers, J.; Worley, K.C.; Aagaard, K.M. Evolutionary genetics and implications of small size and twinning in callitrichine primates. Proc. Natl. Acad. Sci. USA 2014, 111, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.K.; Jones, K.M.; Van Vleck, T.; Emborg, M.E. Modeling genetic diseases in nonhuman primates through embryonic and germline modification: Considerations and challenges. Sci. Transl. Med. 2022, 14, eabf4879. [Google Scholar] [CrossRef] [PubMed]
- Tardif, S.D.; Mansfield, K.G.; Ratnam, R.; Ross, C.N.; Ziegler, T.E. The marmoset as a model of aging and age-related diseases. ILAR J. 2011, 52, 54–65. [Google Scholar] [CrossRef]
- Yun, J.W.; Ahn, J.B.; Kang, B.C. Modeling Parkinson’s disease in the common marmoset (Callithrix jacchus): Overview of models, methods, and animal care. Lab. Anim. Res. 2015, 31, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Lanthier, P.A.; Clark, M.J.; De La Barrera, R.A.; Tighe, M.P.; Szaba, F.M.; Travis, K.L.; Low-Beer, T.C.; Cookenham, T.S.; Lanzer, K.G.; et al. Efficacy of an inactivated Zika vaccine against virus infection during pregnancy in mice and marmosets. NPJ Vaccines 2022, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Singh, B.; Ganatra, S.R.; Gazi, M.; Cole, J.; Thippeshappa, R.; Alfson, K.J.; Clemmons, E.; Gonzalez, O.; Escobedo, R.; et al. Responses to acute infection with SARS-CoV-2 in the lungs of rhesus macaques, baboons and marmosets. Nat. Microbiol. 2021, 6, 73–86. [Google Scholar] [CrossRef]
- Malukiewicz, J.; Boere, V.; De Oliveira, M.A.B.; D’arc, M.; Ferreira, J.V.; French, J.; Housman, G.; De Souza, C.I.; Jerusalinsky, L.; R de Melo, F.; et al. An Introduction to the Callithrix Genus and Overview of Recent Advances in Marmoset Research. ILAR J. 2020, 61, 110–138. [Google Scholar] [CrossRef]
- Miller, C.T.; Freiwald, W.A.; Leopold, D.A.; Mitchell, J.F.; Silva, A.C.; Wang, X. Marmosets: A Neuroscientific Model of Human Social Behavior. Neuron 2016, 90, 219–233. [Google Scholar] [CrossRef]
- Philippens, I.; Langermans, J.A.M. Preclinical Marmoset Model for Targeting Chronic Inflammation as a Strategy to Prevent Alzheimer’s Disease. Vaccines 2021, 9, 388. [Google Scholar] [CrossRef]
- Marmoset Genome, S.; Analysis, C. The common marmoset genome provides insight into primate biology and evolution. Nat. Genet. 2014, 46, 850–857. [Google Scholar] [CrossRef]
- Jayakumar, V.; Ishii, H.; Seki, M.; Kumita, W.; Inoue, T.; Hase, S.; Sato, K.; Okano, H.; Sasaki, E.; Sakakibara, Y. An improved de novo genome assembly of the common marmoset genome yields improved contiguity and increased mapping rates of sequence data. BMC Genom. 2020, 21, 243. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhou, Y.; Marcus, S.; Formenti, G.; Bergeron, L.A.; Song, Z.; Bi, X.; Bergman, J.; Rousselle, M.M.C.; Zhou, C.; et al. Evolutionary and biomedical insights from a marmoset diploid genome assembly. Nature 2021, 594, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Moshiri, A.; Chen, R.; Kim, S.; Harris, R.A.; Li, Y.; Raveendran, M.; Davis, S.; Liang, Q.; Pomerantz, O.; Wang, J.; et al. A nonhuman primate model of inherited retinal disease. J. Clin. Investig. 2019, 129, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Vallender, E.J.; Hotchkiss, C.E.; Lewis, A.D.; Rogers, J.; Stern, J.A.; Peterson, S.M.; Ferguson, B.; Sayers, K. Nonhuman primate genetic models for the study of rare diseases. Orphanet J. Rare Dis. 2023, 18, 20. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Warren, W.C.; Harris, R.A.; Haukness, M.; Fiddes, I.T.; Murali, S.C.; Fernandes, J.; Dishuck, P.C.; Storer, J.M.; Raveendran, M.; Hillier, L.W.; et al. Sequence diversity analyses of an improved rhesus macaque genome enhance its biomedical utility. Science 2020, 370, eabc6617. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Kronenberg, Z.N.; Fiddes, I.T.; Gordon, D.; Murali, S.; Cantsilieris, S.; Meyerson, O.S.; Underwood, J.G.; Nelson, B.J.; Chaisson, M.J.P.; Dougherty, M.L.; et al. High-resolution comparative analysis of great ape genomes. Science 2018, 360, eaar6343. [Google Scholar] [CrossRef]
- Sweeney, C.G.; Curran, E.; Westmoreland, S.V.; Mansfield, K.G.; Vallender, E.J. Quantitative molecular assessment of chimerism across tissues in marmosets and tamarins. BMC Genom. 2012, 13, 98. [Google Scholar] [CrossRef]
- Reid, J.G.; Carroll, A.; Veeraraghavan, N.; Dahdouli, M.; Sundquist, A.; English, A.; Bainbridge, M.; White, S.; Salerno, W.; Buhay, C.; et al. Launching genomics into the cloud: Deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinform. 2014, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Coe, B.P.; Stessman, H.A.F.; Sulovari, A.; Geisheker, M.R.; Bakken, T.E.; Lake, A.M.; Dougherty, J.D.; Lein, E.S.; Hormozdiari, F.; Bernier, R.A.; et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat. Genet. 2019, 51, 106–116. [Google Scholar] [CrossRef]
- Stage, E.; Risacher, S.L.; Lane, K.A.; Gao, S.; Nho, K.; Saykin, A.J.; Apostolova, L.G.; Alzheimer’s Disease Neuroimaging Initiative. Association of the top 20 Alzheimer’s disease risk genes with [(18)F]flortaucipir PET. Alzheimers Dement. 2022, 14, e12308. [Google Scholar] [CrossRef]
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Raney, B.J.; Dreszer, T.R.; Barber, G.P.; Clawson, H.; Fujita, P.A.; Wang, T.; Nguyen, N.; Paten, B.; Zweig, A.S.; Karolchik, D.; et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics 2014, 30, 1003–1005. [Google Scholar] [CrossRef]
- Tapmeier, T.T.; Rahmioglu, N.; Lin, J.; De Leo, B.; Obendorf, M.; Raveendran, M.; Fischer, O.M.; Bafligil, C.; Guo, M.; Harris, R.A.; et al. Neuropeptide S receptor 1 is a nonhormonal treatment target in endometriosis. Sci. Transl. Med. 2021, 13, eabd6469. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.M.; McGill, T.J.; Puthussery, T.; Stoddard, J.; Renner, L.; Lewis, A.D.; Colgin, L.M.A.; Gayet, J.; Wang, X.; Prongay, K.; et al. Bardet-Biedl Syndrome in rhesus macaques: A nonhuman primate model of retinitis pigmentosa. Exp. Eye Res. 2019, 189, 107825. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, J.N.; Ross, C.N.; Ziegler, T.; Burke, L.A.; Steffen, A.D.; Sills, A.; Layne Colon, D.; de Martelly, V.A.; Narapareddy, L.R.; Tardif, S.D. Womb to womb: Maternal litter size and birth weight but not adult characteristics predict early neonatal death of offspring in the common marmoset monkey. PLoS ONE 2021, 16, e0252093. [Google Scholar] [CrossRef] [PubMed]
- Power, M.L.; Ross, C.N.; Schulkin, J.; Ziegler, T.E.; Tardif, S.D. Metabolic consequences of the early onset of obesity in common marmoset monkeys. Obesity 2013, 21, E592–E598. [Google Scholar] [CrossRef]
- Zoratto, F.; Sinclair, E.; Manciocco, A.; Vitale, A.; Laviola, G.; Adriani, W. Individual differences in gambling proneness among rats and common marmosets: An automated choice task. BioMed Res. Int. 2014, 2014, 927685. [Google Scholar] [CrossRef]
- Ash, H.; Chang, A.; Ortiz, R.J.; Kulkarni, P.; Rauch, B.; Colman, R.; Ferris, C.F.; Ziegler, T.E. Structural and functional variations in the prefrontal cortex are associated with learning in pre-adolescent common marmosets (Callithrix jacchus). Behav. Brain Res. 2022, 430, 113920. [Google Scholar] [CrossRef]
- Ash, H.; Ziegler, T.E.; Colman, R.J. Early learning in the common marmoset (Callithrix jacchus): Behavior in the family group is related to preadolescent cognitive performance. Am. J. Primatol. 2020, 82, e23159. [Google Scholar] [CrossRef] [PubMed]
- Vermilyea, S.C.; Babinski, A.; Tran, N.; To, S.; Guthrie, S.; Kluss, J.H.; Schmidt, J.K.; Wiepz, G.J.; Meyer, M.G.; Murphy, M.E.; et al. In Vitro CRISPR/Cas9-Directed Gene Editing to Model LRRK2 G2019S Parkinson’s Disease in Common Marmosets. Sci. Rep. 2020, 10, 3447. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.; Lyke, M.; Christensen, C.; Newman, D.; Bagwell, A.; Galindo, S.; Glenn, J.; Layne-Colon, D.; Sayers, K.; Tardif, S.; et al. Genetic characterization of a captive marmoset colony using genotype-by-sequencing. bioRxiv 2023. [Google Scholar] [CrossRef]
- Rautiainen, M.; Nurk, S.; Walenz, B.P.; Logsdon, G.A.; Porubsky, D.; Rhie, A.; Eichler, E.E.; Phillippy, A.M.; Koren, S. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. 2023, 41, 1474–1482. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Marmoset SNV ID | dbSNP | Gene | Marmoset VEP Consequence | Allele Freq. | ClinVar Disease |
|---|---|---|---|---|---|
| Pathogenic | |||||
| 13:7334434:C:T | rs864321699 | GATA4 | Missense | 0.0179 | Congenital heart disease |
| 15:3504507:T:C | rs577069249 | CCDC39 | Intron | 0.1369 | Primary ciliary dyskinesia |
| 16:51588847:C:T | rs994355873 | RECQL4 | Stop gained | 0.0060 | Baller–Gerold syndrome |
| 19:10341384:C:T | rs770329105 | USH2A | Missense | 0.0833 | Usher syndrome type 2A |
| 22:39820446:C:T | rs80356463 | SIX5 | Missense | 0.0060 | Branchiootorenal syndrome 2 |
| 2:44720125:C:A | rs191068215 | SPINK1 | 5 prime UTR | 0.0119 | Tropical pancreatitis |
| 4:17947215:G:A | rs104893960 | GCM2 | Missense | 0.0357 | Hypoparathyroidism, familial isolated, 2 |
| 5:96794138:G:A | rs764561297 | HNF1B | Synonymous | 0.0238 | Maturity onset diabetes mellitus in young |
| 7:149491445:G:A | rs606231300 | SLC16A1 | Stop gained | 0.2321 | Monocarboxylate transporter 1 deficiency, autosomal dominant |
| 9:68087733:C:T | rs1942608745 | KMT2D | Stop gained | 0.3988 | Kabuki syndrome |
| 9:68088117:C:T | rs1942584412 | KMT2D | Missense | 0.3988 | Kabuki syndrome |
| Pathogenic/Likely Pathogenic | |||||
| 14:36823751:C:T | rs1057521141 | DYSF | Missense | 0.0238 | Autosomal recessive limb-girdle muscular dystrophy type 2B/qualitative or quantitative defects of dysferlin |
| 5:65779382:G:A | rs104894553 | ASPA | Missense | 0.5000 | Spongy degeneration of the central nervous system/mild Canavan disease/inborn genetic diseases |
| 9:68087587:G:A | rs886041404 | KMT2D | Missense | 0.3976 | Kabuki syndrome |
| 9:68087980:G:A | rs267607237 | KMT2D | Missense | 0.3988 | Kabuki syndrome |
| Likely Pathogenic | |||||
| 10:107534744:C:T | rs751122392 | IFT43 | Intron | 0.0179 | Not provided |
| 10:42430818:C:T | rs979186313 | KNL1 | Synonymous | 0.7917 | Microcephaly 4, primary, autosomal recessive |
| 11:5831070:A:G | rs864622543 | ATM | Intron | 0.4702 | Ataxia–telangiectasia syndrome/hereditary cancer-predisposing syndrome |
| 18:26568817:T:C | rs118203910 | F5 | Missense | 0.0238 | Factor V deficiency |
| 2:1601055:G:T | rs200853731 | ASL | Missense | 0.0119 | Argininosuccinate lyase deficiency |
| 4:46330371:C:T | rs2113877345 | TREM2 | Splice acceptor | 0.0119 | Not provided |
| 4:57040110:C:A | rs2128209217 | PKHD1 | Splice donor | 0.7440 | Autosomal recessive polycystic kidney disease |
| 5:10531420:T:C | rs576243101 | PROKR2 | Missense | 0.3155 | Not provided |
| 5:36862718:G:A | rs35187177 | SGK2 | Missense | 0.0179 | Colorectal cancer |
| 5:5037801:C:T | rs748725549 | XKR7 | Missense | 0.0119 | Moyamoya angiopathy |
| 9:68087755:G:T | rs2120361380 | KMT2D | Missense | 0.3988 | Kabuki syndrome 1 |
| 9:90628336:C:T | rs1381940328 | CEP290 | Intron | 0.1905 | Leber congenital amaurosis 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harris, R.A.; Raveendran, M.; Warren, W.; LaDeana, H.W.; Tomlinson, C.; Graves-Lindsay, T.; Green, R.E.; Schmidt, J.K.; Colwell, J.C.; Makulec, A.T.; et al. Whole Genome Analysis of SNV and Indel Polymorphism in Common Marmosets (Callithrix jacchus). Genes 2023, 14, 2185. https://doi.org/10.3390/genes14122185
Harris RA, Raveendran M, Warren W, LaDeana HW, Tomlinson C, Graves-Lindsay T, Green RE, Schmidt JK, Colwell JC, Makulec AT, et al. Whole Genome Analysis of SNV and Indel Polymorphism in Common Marmosets (Callithrix jacchus). Genes. 2023; 14(12):2185. https://doi.org/10.3390/genes14122185
Chicago/Turabian StyleHarris, R. Alan, Muthuswamy Raveendran, Wes Warren, Hillier W. LaDeana, Chad Tomlinson, Tina Graves-Lindsay, Richard E. Green, Jenna K. Schmidt, Julia C. Colwell, Allison T. Makulec, and et al. 2023. "Whole Genome Analysis of SNV and Indel Polymorphism in Common Marmosets (Callithrix jacchus)" Genes 14, no. 12: 2185. https://doi.org/10.3390/genes14122185
APA StyleHarris, R. A., Raveendran, M., Warren, W., LaDeana, H. W., Tomlinson, C., Graves-Lindsay, T., Green, R. E., Schmidt, J. K., Colwell, J. C., Makulec, A. T., Cole, S. A., Cheeseman, I. H., Ross, C. N., Capuano, S., III, Eichler, E. E., Levine, J. E., & Rogers, J. (2023). Whole Genome Analysis of SNV and Indel Polymorphism in Common Marmosets (Callithrix jacchus). Genes, 14(12), 2185. https://doi.org/10.3390/genes14122185