
Variable Selection for Sparse Data with Applications to Vaginal Microbiome and Gene Expression Data



Abstract
:1. Introduction
2. Materials and Methods
2.1. Zero-Altered or Hurdle Models
2.2. Zero-Inflated Models
2.3. Zero-Altered and Zero-Inflated Models with Continuous Baseline Distributions
2.4. Model Selection Using AZIAD Package
2.5. Significance Test on Group Labels
- Step 1: Choose the most appropriate model for all the N numbers, after ignoring their class labels. This task is accomplished by performing KS-tests using kstest.A on all models under consideration (see also [7]). Then we compute the MLE of the parameters for the chosen model using R function . The corresponding AIC value is denoted by .
- Step 2: For each of the m classes, say the kth class, we choose the most appropriate model for the data of the kth class, compute the MLE and denote the corresponding AIC value by . Then aggregated AIC value is essentially the summation of the AIC values from m classes, that is, .
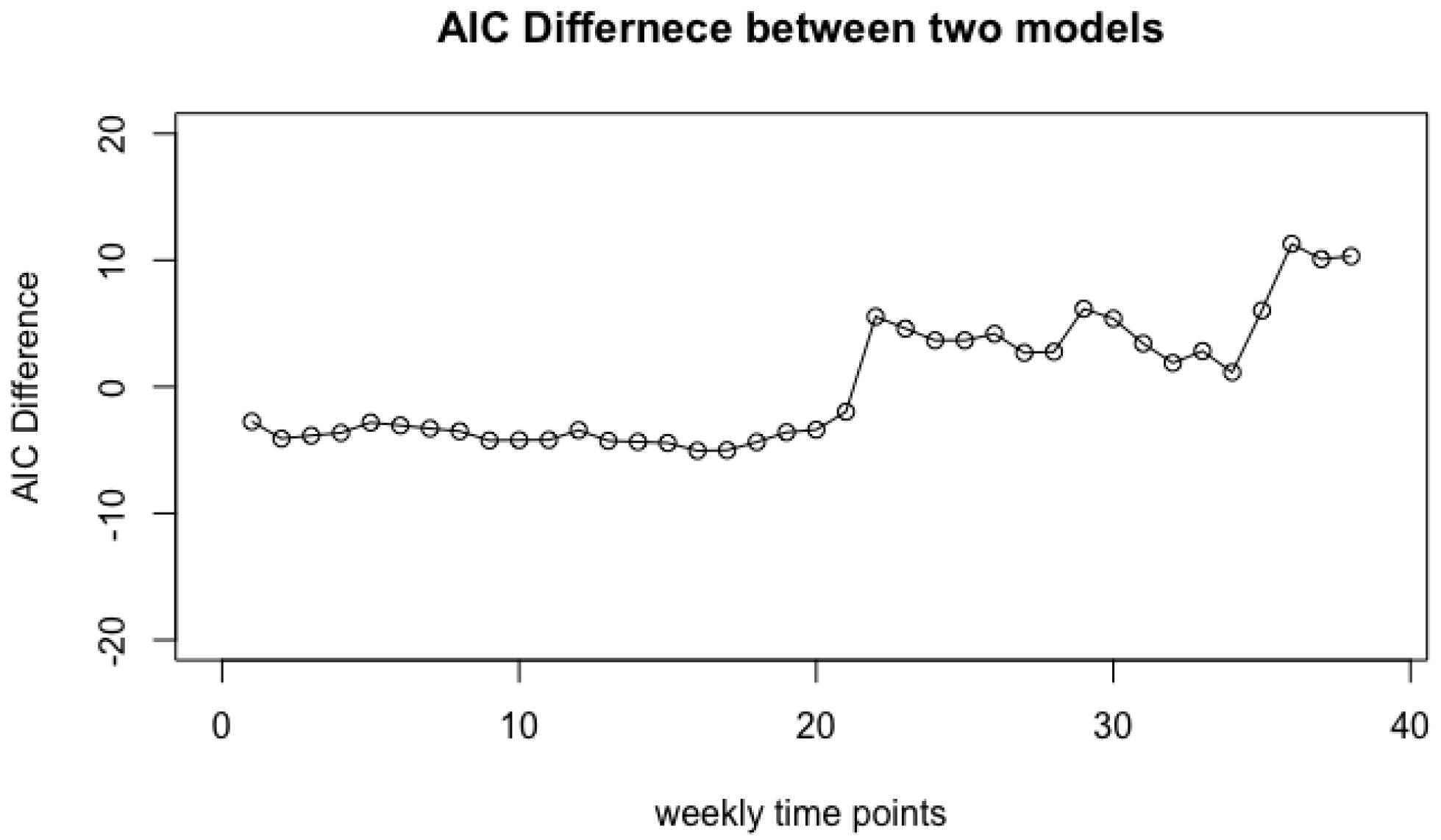
- Step 3: Take the difference of two AIC values with or without class labels, . A larger difference indicates that the jth covariate is more informative for predicting the class labels.
3. Two Applications
3.1. Vaginal Microbiome
3.2. RNA-Seq Gene Expression Data
4. Data Analysis and Results
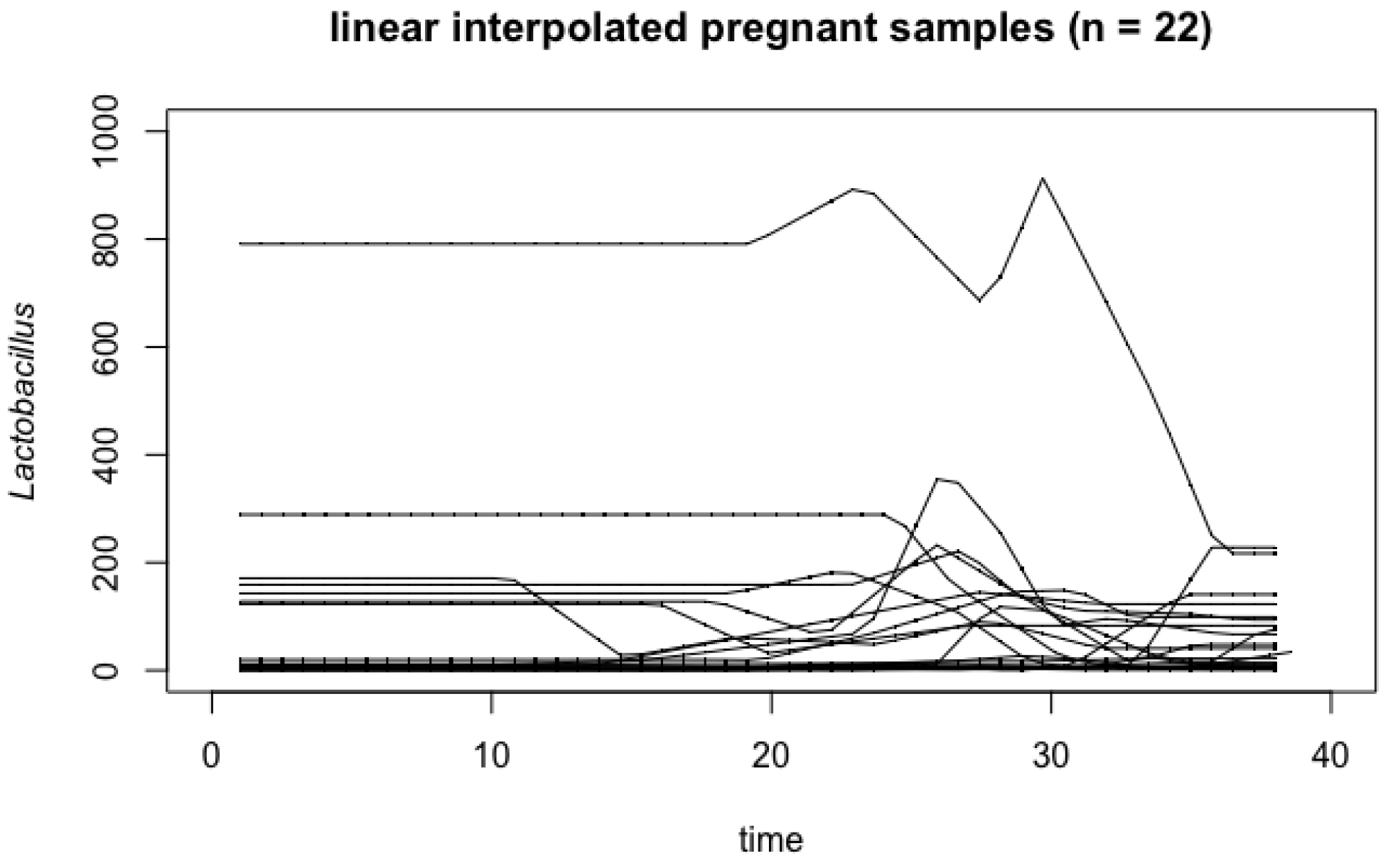
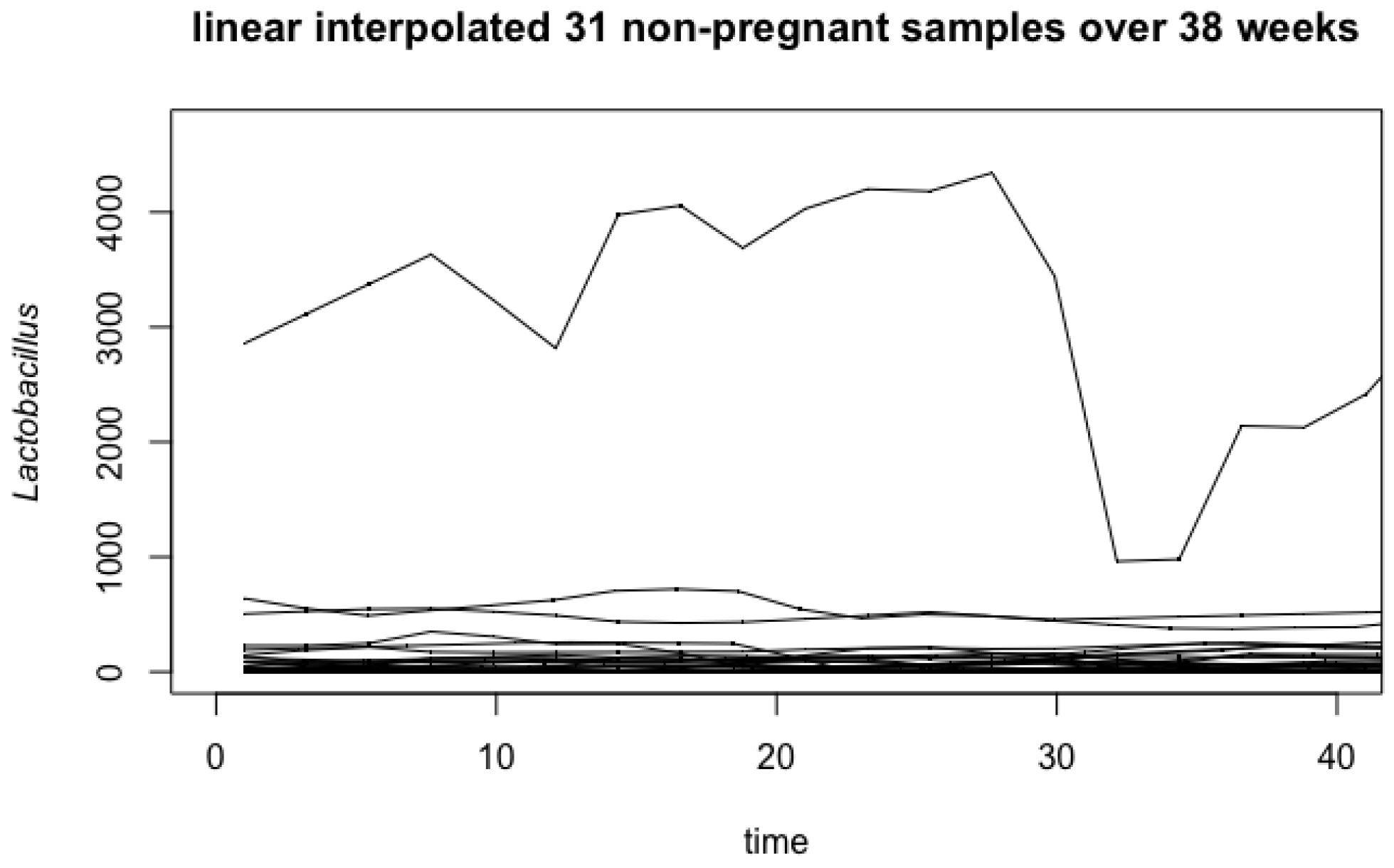
4.1. Vaginal Microbiome
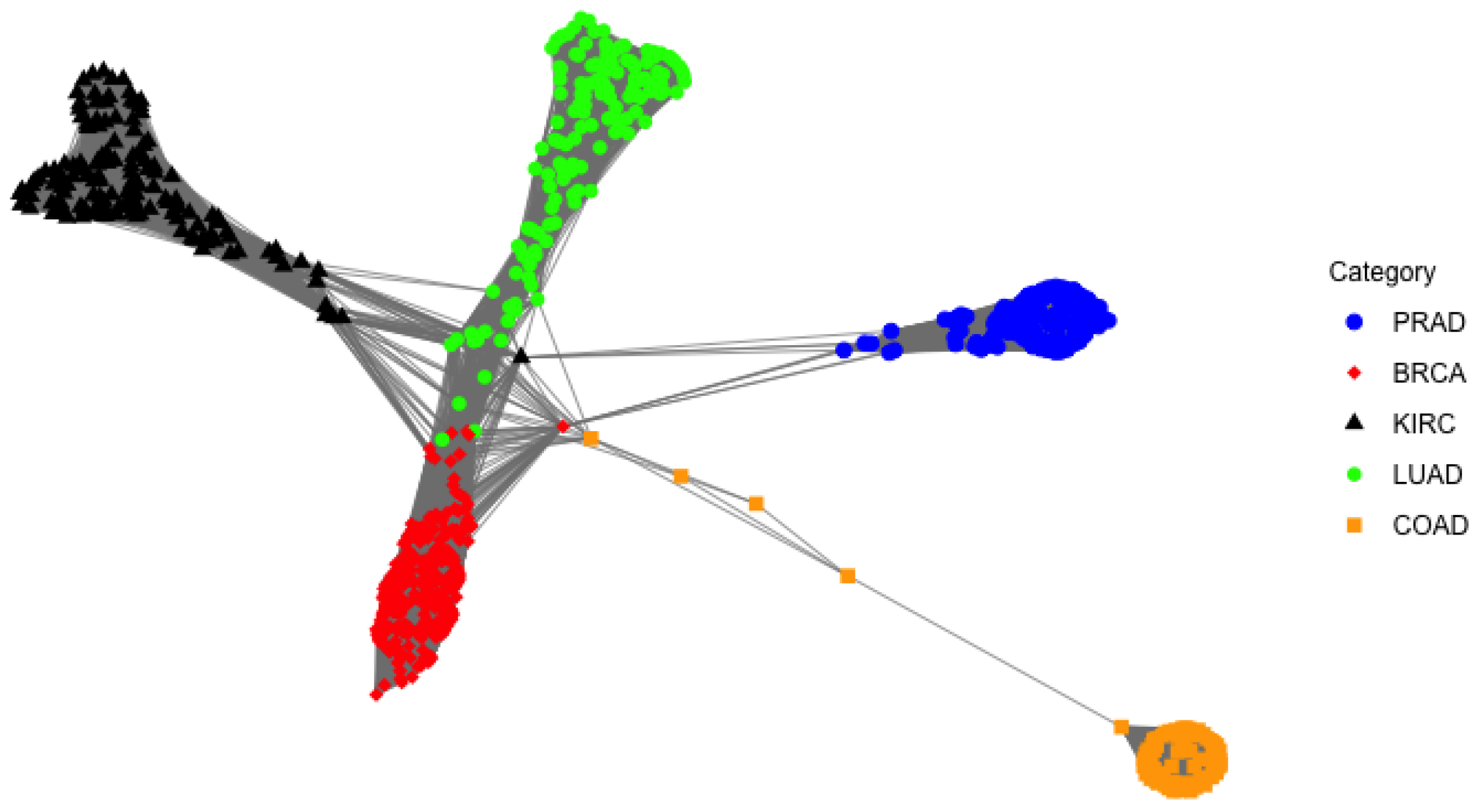
4.2. RNA-Seq Gene Expression Data
5. Conclusions and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Influence of Outlier in Vaginal Microbiome Analysis

Appendix B. More KS-Test Results for Vaginal Microbiome Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (Week) | N | ZIN | NH | HN | ZIHN | HNH | LN | ZILN | LNH | E | ZIE | EH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| week 10 | 1.000 | 0.395 | 1.000 | 0.460 | 0.580 | 0.020 | 0.000 | 0.000 | 0.870 | 0.330 | 0.725 | 0.040 |
| week 22 | 1.000 | 0.480 | 1.000 | 0.520 | 0.420 | 0.010 | 0.000 | 0.000 | 0.785 | 0.450 | 0.590 | 0.025 |
| week 36 | 1.000 | 0.160 | 1.000 | 0.480 | 0.790 | 0.020 | 0.000 | 0.000 | 0.955 | 0.145 | 0.375 | 0.015 |
| Time (Week) | N | ZIN | NH | HN | ZIHN | HNH | LN | ZILN | LNH | E | ZIE | EH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| week 10 | 1.000 | 0.355 | 1.000 | 0.505 | 0.385 | 0.170 | 0.000 | 0.000 | 0.390 | 0.500 | 0.130 | 0.130 |
| week 22 | 1.000 | 0.250 | 1.000 | 0.295 | 0.490 | 0.415 | 0.020 | 0.000 | 0.000 | 0.135 | 0.005 | 0.170 |
| week 36 | 1.000 | 0.380 | 1.000 | 0.635 | 0.975 | 0.560 | 0.005 | 0.000 | 0.000 | 0.425 | 0.550 | 0.410 |
Appendix C. List of 50 Selected Genes for Gene Expression Data
References
- Metwally, A.A.; Aldirawi, H.; Yang, J. A review on probabilistic models used in microbiome studies. Commun. Inf. Syst. 2018, 18, 173–191. [Google Scholar] [CrossRef]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Nikita, L.; Galuppi, M.; Lamont, R.F.; Chaemsaithong, P.; Miranda, J.; et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Pati, D.; Mallick, B.K.; Carroll, R.J. Bayesian copula density deconvolution for zero-inflated data in nutritional epidemiology. J. Am. Stat. Assoc. 2021, 116, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Aljabri, D.; Vaughn, A.; Austin, M.; White, L.; Li, Z.; Naessens, J.; Spaulding, A. An investigation of healthcare worker perception of their workplace safety and incidence of injury. Workplace Health Saf. 2020, 68, 214–225. [Google Scholar] [CrossRef]
- Chen, P.; Liu, Q.; Sun, F. Bicycle parking security and built environments. Transp. Res. Part D Transp. Environ. 2018, 62, 169–178. [Google Scholar] [CrossRef]
- Kim, A. Social exclusion of multicultural families in Korea. Soc. Sci. 2018, 7, 63. [Google Scholar] [CrossRef]
- Aldirawi, H.; Yang, J.; Metwally, A.A. Identifying Appropriate Probabilistic Models for Sparse Discrete Omics Data. In Proceedings of the 2019 IEEE EMBS International Conference on Biomedical & Health Informatics (BHI), Chicago, IL, USA, 19–22 May 2019; IEEE: Piscataway, NJ, USA, 2019; pp. 1–4. [Google Scholar]
- Aldirawi, H.; Yang, J. Modeling Sparse Data Using MLE with Applications to Microbiome Data. J. Stat. Theory Pract. 2022, 16, 13. [Google Scholar] [CrossRef]
- Jiang, R.; Sun, T.; Song, D.; Li, J.J. Statistics or biology: The zero-inflation controversy about scRNA-seq data. Genome Biol. 2022, 23, 1–24. [Google Scholar] [CrossRef]
- Lambert, D. Zero-inflated Poisson regression, with an application to defects in manufacturing. Technometrics 1992, 34, 1–14. [Google Scholar] [CrossRef]
- Greene, W.H. Accounting for Excess Zeros and Sample Selection in Poisson and Negative Binomial Regression Models. NYU 30 Working Paper No. EC-94-10. 1994. Available online: https://ssrn.com/abstract=1293115 (accessed on 5 November 2022). NYU Working Paper No. EC-94-10.
- Reid, G.; Bocking, A. The potential for probiotics to prevent bacterial vaginosis and preterm labor. Am. J. Obstet. Gynecol. 2003, 189, 1202–1208. [Google Scholar] [CrossRef]
- Witkin, S.S.; Linhares, I.M. Why do lactobacilli dominate the human vaginal microbiota? BJOG Int. J. Obstet. Gynaecol. 2017, 124, 606–611. [Google Scholar] [CrossRef]
- Eschenbach, D.A.; Davick, P.R.; Williams, B.L.; Klebanoff, S.J.; Young-Smith, K.; Critchlow, C.M.; Holmes, K.K. Prevalence of hydrogen peroxide-producing Lactobacillus species in normal women and women with bacterial vaginosis. J. Clin. Microbiol. 1989, 27, 251–256. [Google Scholar] [CrossRef]
- Hawes, S.E.; Hillier, S.L.; Benedetti, J.; Stevens, C.E.; Koutsky, L.A.; Wølner-Hanssen, P.; Holmes, K.K. Hydrogen peroxide—Producing lactobacilli and acquisition of vaginal infections. J. Infect. Dis. 1996, 174, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Klaenhammer, T.R. Bacteriocins of lactic acid bacteria. Biochimie 1988, 70, 337–349. [Google Scholar] [CrossRef]
- Ng, S.; Hart, A.; Kamm, M.; Stagg, A.; Knight, S.C. Mechanisms of action of probiotics: Recent advances. Inflamm. Bowel Dis. 2009, 15, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Koedooder, R.; Singer, M.; Schoenmakers, S.; Savelkoul, P.H.; Morré, S.A.; de Jonge, J.D.; Poort, L.; Cuypers, W.J.S.; Beckers, N.; Broekmans, F.; et al. The vaginal microbiome as a predictor for outcome of in vitro fertilization with or without intracytoplasmic sperm injection: A prospective study. Hum. Reprod. 2019, 34, 1042–1054. [Google Scholar] [CrossRef]
- Chen, E.Z.; Li, H. A two-part mixed-effects model for analyzing longitudinal microbiome compositional data. Bioinformatics 2016, 32, 2611–2617. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, B.; Yi, N. Zero-inflated Gaussian mixed models for analyzing longitudinal microbiome data. PLoS ONE 2020, 15, e0242073. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.W.; He, Q.; Huang, H.H. Clustering Gene Expressions Using the Table Invitation Prior. Genes 2022, 13, 2036. [Google Scholar] [CrossRef]
- Ahlmann-Eltze, C.; Huber, W. glmGamPoi: Fitting Gamma-Poisson generalized linear models on single cell count data. Bioinformatics 2020, 36, 5701–5702. [Google Scholar] [CrossRef]
- Ji, F.; Sadreyev, R.I. RNA-seq: Basic bioinformatics analysis. Curr. Protoc. Mol. Biol. 2018, 124, e68. [Google Scholar] [CrossRef] [PubMed]
- Zappia, L.; Phipson, B.; Oshlack, A. Splatter: Simulation of single-cell RNA sequencing data. Genome Biol. 2017, 18, 174. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 2014, 11, 740–742. [Google Scholar] [CrossRef] [PubMed]
- McDavid, A.; Finak, G.; Chattopadyay, P.K.; Dominguez, M.; Lamoreaux, L.; Ma, S.S.; Roederer, M.; Gottardo, R. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics 2013, 29, 461–467. [Google Scholar] [CrossRef]
- Peng, X.; Li, G.; Liu, Z. Zero-inflated beta regression for differential abundance analysis with metagenomics data. J. Comput. Biol. 2016, 23, 102–110. [Google Scholar] [CrossRef]
- Cho, H.; Liu, C.; Park, J.; Wu, D. bzinb: Bivariate Zero-Inflated Negative Binomial Model Estimator; R Package Version 1.0.4; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Balderama, E.; Trippe, T. hurdlr: Zero-Inflated and Hurdle Modelling Using Bayesian Inference; R Package Version 0.1; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Wang, L.; Aldirawi, H.; Yang, J. iZID: Identify Zero-Inflated Distributions; R Package Version 0.0.1; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Stasinopoulos, M. gamlss: Generalised Additive Models for Location Scale and Shape; R Package Version 0.0.1; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Jackman, S. pscl: Political Science Computational Laboratory; R Package Version 0.0.1; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Croissant, Y.; Carlevaro, F.; Hoareau, S. mhurdle: Multiple Hurdle Tobit Models; R Package Version 1.3.0; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Waudby-Smith, I.; Li, P. rbtt: Alternative Bootstrap-Based t-Test Aiming to Reduce Type-I Error for Non-Negative, Zero-Inflated Data; R Package Version 0.1.0; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Peng, X.; Li, G.; Liu, Z.; Chen, H. ZIBseq: Differential Abundance Analysis for Metagenomic Data via Zero-Inflated Beta Regression; R Package Version 1.2; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Jochmann, M. zic: Bayesian Inference for Zero-Inflated Count Models; R Package Version 0.9.1; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Yang, M.; Zamba, G.; Cavanaugh, J. ZIM: Zero-Inflated Models (ZIM) for Count Time Series with Excess Zeros; R Package Version 1.1.0; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Xu, Z.J.; Liu, Y. ziphsmm: Zero-Inflated Poisson Hidden (Semi-)Markov Models; R Package Version 2.0.6; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Wang, L.; Aldirawi, H.; Yang, J. Identifying zero-inflated distributions with a new R package iZID. Commun. Inf. Syst. 2020, 20, 23–44. [Google Scholar] [CrossRef]
- Dousti Mousavi, N.; Aldirawi, H.; Yang, J. AZIAD: Analyzing Zero-Inflated and Zero-Altered Data; R Package Version 0.0.2; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Dousti Mousavi, N.; Aldirawi, H.; Yang, J. An R Package AZIAD for Analyzing Zero-Inflated and Zero-Altered Data. arXiv 2022, arXiv:2205.01294. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Burnham, K.P.; Anderson, D.R. Multimodel inference: Understanding AIC and BIC in model selection. Sociol. Methods Res. 2004, 33, 261–304. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Gelman, A.; Hill, J. Data Analysis Using Regression and Multilevel/Hierarchical Models; Analytical Methods for Social Research; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar] [CrossRef]
- Metwally, A.A.; Yang, J.; Ascoli, C.; Dai, Y.; Finn, P.W.; Perkins, D.L. MetaLonDA: A flexible R package for identifying time intervals of differentially abundant features in metagenomic longitudinal studies. Microbiome 2018, 6, 32. [Google Scholar] [CrossRef]
- Harrison, C.W.; He, Q.; Huang, H.H. tip: Bayesian Clustering Using the Table Invitation Prior (TIP); R Package Version 0.1.0; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]







| Time (Week) | N | ZIN | NH | HN | ZIHN | HNH | LN | ZILN | LNH | E | ZIE | EH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| week 10 | 1.000 | 0.390 | 1.000 | 0.315 | 0.510 | 0.000 | 0.000 | 0.000 | 0.720 | 0.335 | 0.510 | 0.015 |
| week 22 | 1.000 | 0.160 | 1.000 | 0.605 | 0.625 | 0.000 | 0.000 | 0.000 | 0.955 | 0.100 | 0.300 | 0.020 |
| week 36 | 1.000 | 0.175 | 1.000 | 0.560 | 0.650 | 0.015 | 0.000 | 0.000 | 0.960 | 0.525 | 0.450 | 0.030 |
| Number of Genes | Prediction Error |
|---|---|
| 20 | |
| 50 | |
| 100 | |
| 2426 | |
| Number of Genes | Prediction Error |
|---|---|
| 10 | |
| 20 | |
| 30 | |
| 40 | |
| 50 | 0 |
| 60 | |
| 100 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dousti Mousavi, N.; Yang, J.; Aldirawi, H. Variable Selection for Sparse Data with Applications to Vaginal Microbiome and Gene Expression Data. Genes 2023, 14, 403. https://doi.org/10.3390/genes14020403
Dousti Mousavi N, Yang J, Aldirawi H. Variable Selection for Sparse Data with Applications to Vaginal Microbiome and Gene Expression Data. Genes. 2023; 14(2):403. https://doi.org/10.3390/genes14020403
Chicago/Turabian StyleDousti Mousavi, Niloufar, Jie Yang, and Hani Aldirawi. 2023. "Variable Selection for Sparse Data with Applications to Vaginal Microbiome and Gene Expression Data" Genes 14, no. 2: 403. https://doi.org/10.3390/genes14020403
APA StyleDousti Mousavi, N., Yang, J., & Aldirawi, H. (2023). Variable Selection for Sparse Data with Applications to Vaginal Microbiome and Gene Expression Data. Genes, 14(2), 403. https://doi.org/10.3390/genes14020403