
The Complete Mitochondrial Genome of Dendrogale murina (Tupaiidae) and Phylogeny of Scandentia

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
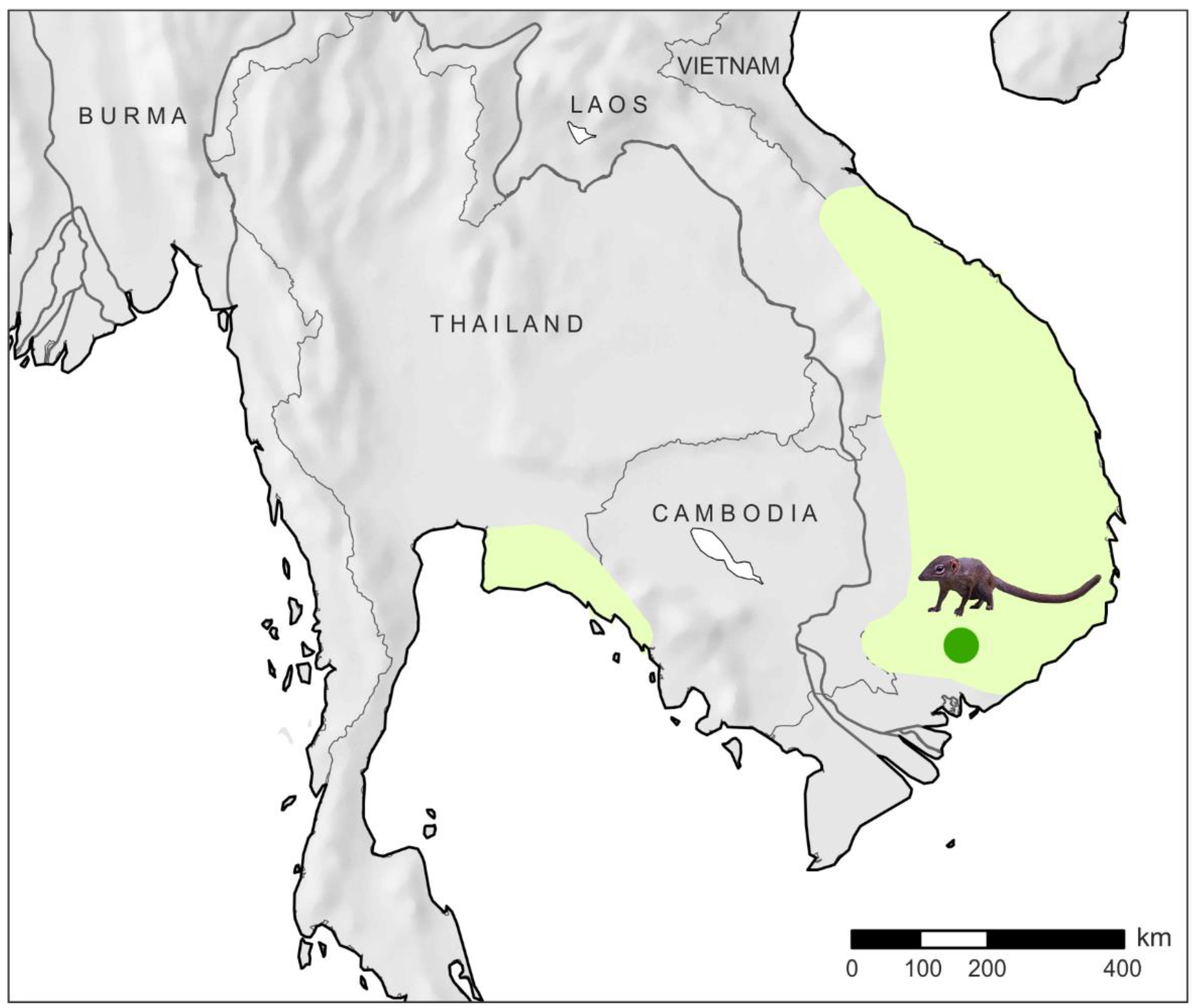
2.1. Material
2.2. DNA Extraction, Library Preparation, and Sequencing
2.3. Mitochondrial Genome Assembly, Annotation, and Sequence Analyses
2.4. Phylogenetic Reconstruction and Divergence Time Estimation
3. Results
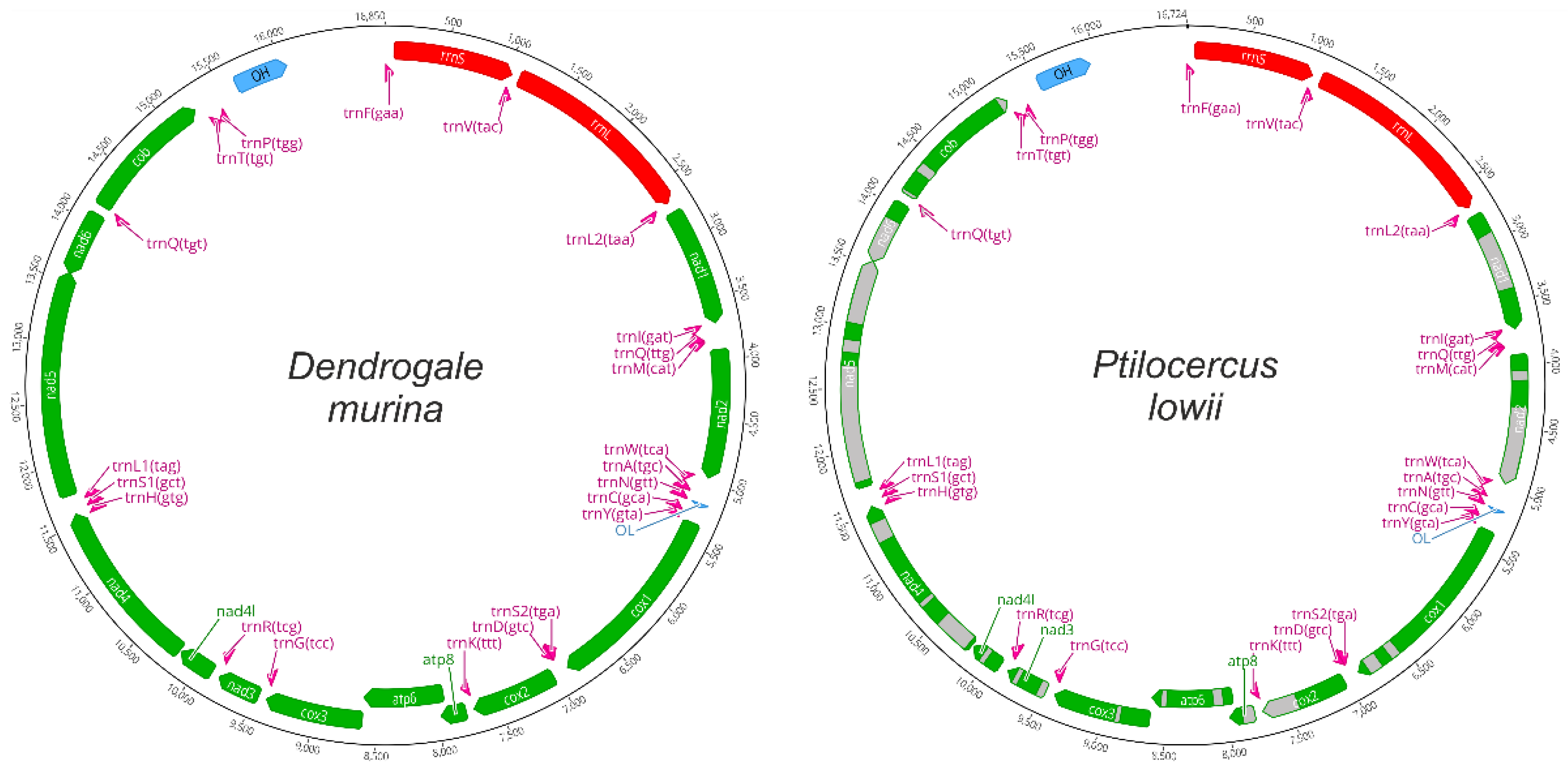
3.1. Mitochondrial Genome Structure and Composition
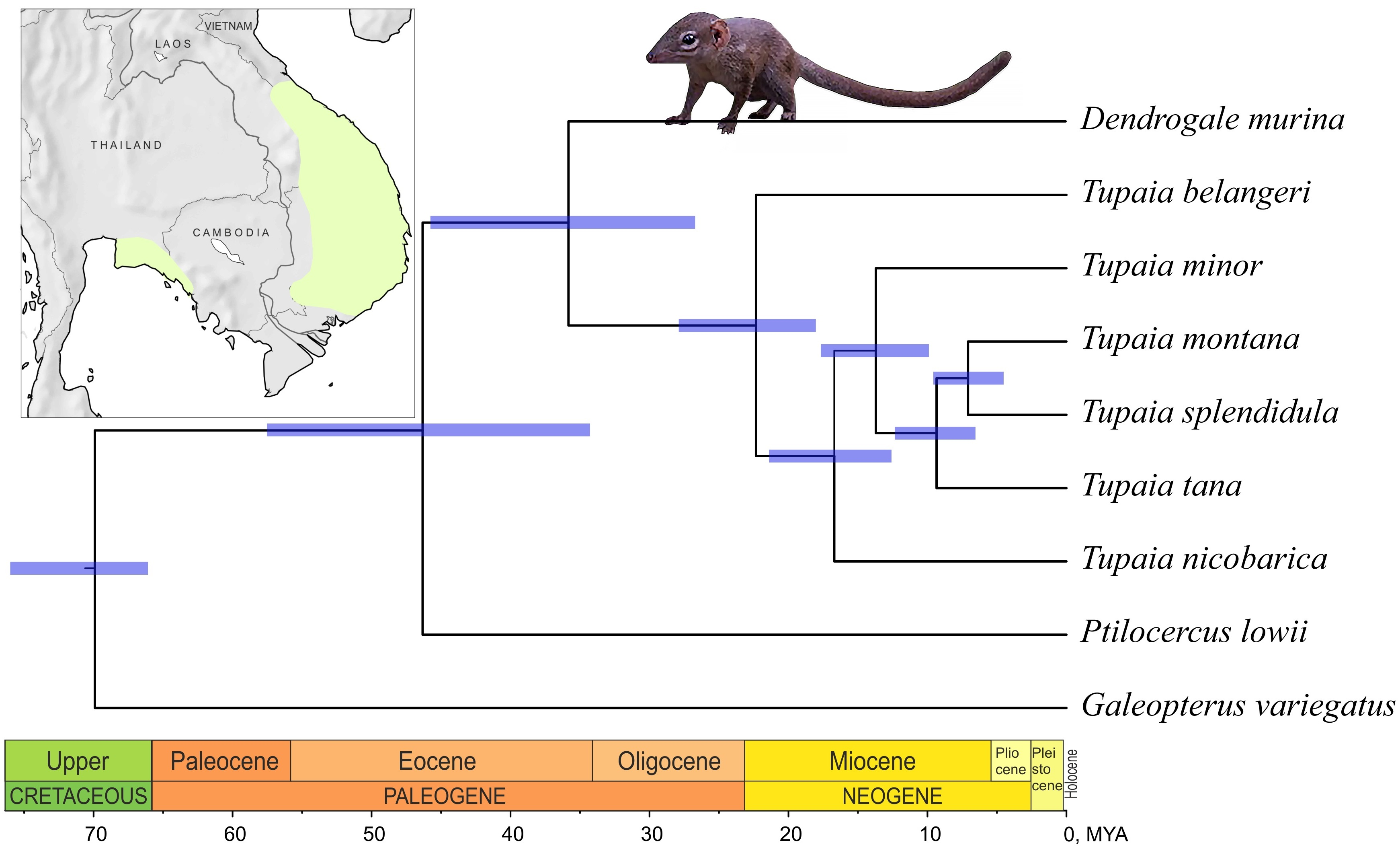
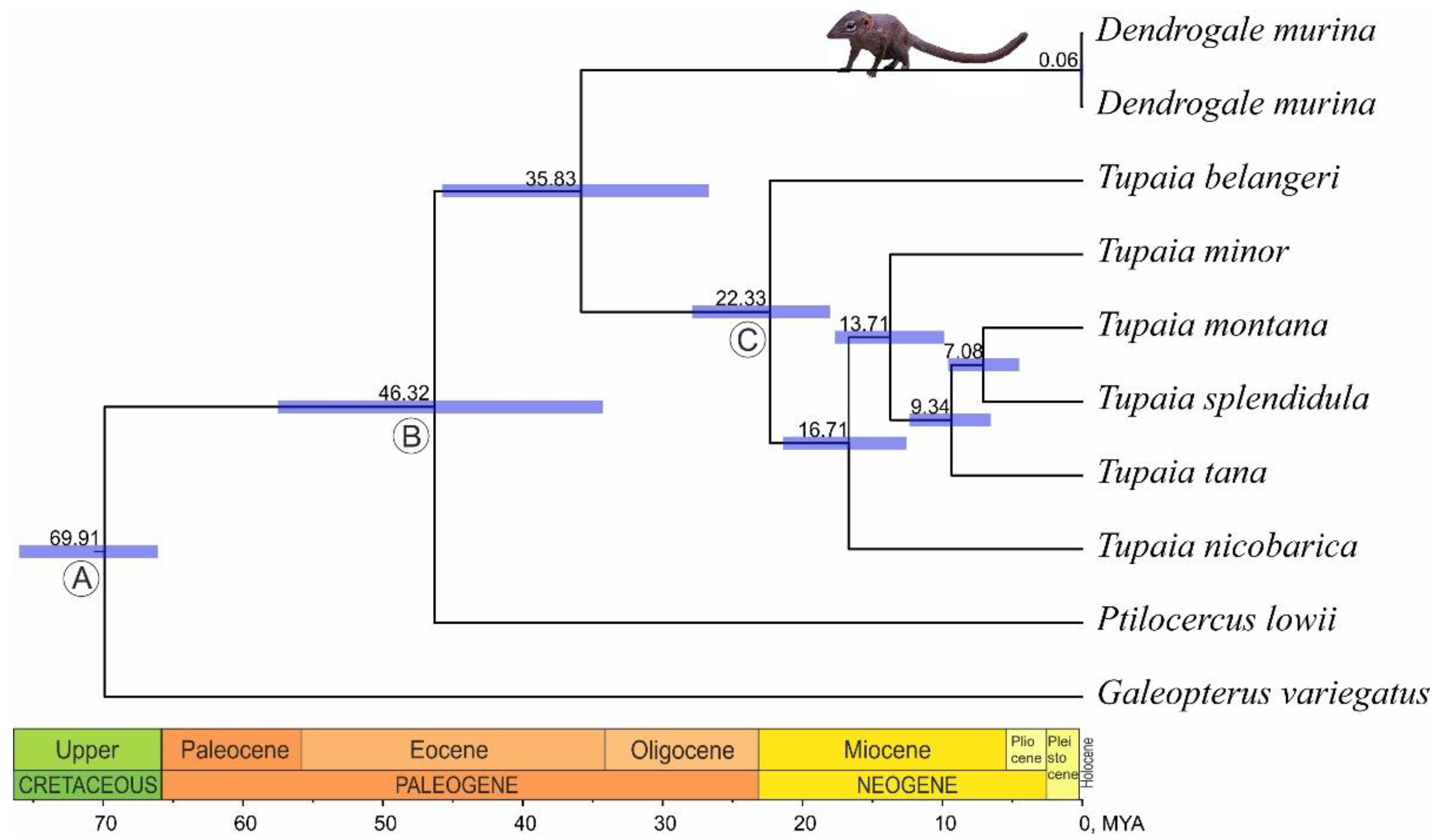
3.2. Phylogenetic Reconstruction and Divergence Time Estimation
4. Discussion
4.1. New Mitochondrial Genomes Assembled
4.2. The Phylogenetic Relationships of Genera within the Order and Time Estimates
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martin, R.D. Primate Origins and Evolution; Princeton University Press: Princeton, NJ, USA, 1990. [Google Scholar]
- MacPhee, R.D.E. (Ed.) Primates and Their Relatives in Phylogenetic Perspective; Plenum Press: New York, NY, USA, 1993. [Google Scholar]
- Murphy, W.J.; Eizirik, E.; Johnson, W.E.; Zhang, Y.P.; Ryder, O.A.; O’Brien, S.J. Molecular Phylogenetics and the Origins of Placental Mammals. Nature 2001, 409, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Silcox, M.T. A Phylogenetic Analysis of Plesiadapiformes and Their Relationship to Euprimates and Other Archontans. Ph.D. Thesis, Johns Hopkins University School of Medicine, Baltimore, MD, USA, 2001. [Google Scholar]
- Bloch, J.I.; Boyer, D.M. Grasping Primate Origins. Science 2002, 298, 1606–1610. [Google Scholar] [CrossRef] [Green Version]
- Sargis, E.J. A Preliminary Qualitative Analysis of the Axial Skeleton of Tupaiids (Mammalia, Scandentia): Functional Morphology and Phylogenetic Implications. J. Zool. 2001, 253, 473–483. [Google Scholar] [CrossRef]
- Sargis, E.J. Functional Morphology of the Forelimb of Tupaiids (Mammalia, Scandentia) and Its Phylogenetic Implications. J. Morphol. 2002, 253, 10–42. [Google Scholar] [CrossRef]
- Sargis, E.J. Functional Morphology of the Hindlimb of Tupaiids (Mammalia, Scandentia) and Its Phylogenetic Implications. J. Morphol. 2002, 254, 149–185. [Google Scholar] [CrossRef]
- Sargis, E.J. The Postcranial Morphology of Ptilocercus Lowii (Scandentia, Tupaiidae): An Analysis of Primatomorphan and Volitantian Characters. J. Mamm. Evol. 2002, 9, 137–160. [Google Scholar] [CrossRef]
- Olson, L.E.; Sargis, E.J.; Martin, R.D. Phylogenetic Relationships among Treeshrews (Scandentia): A Review and Critique of the Morphological Evidence. J. Mamm. Evol. 2004, 11, 49–71. [Google Scholar] [CrossRef]
- Roberts, T.E.; Lanier, H.C.; Sargis, E.J.; Olson, L.E. Molecular Phylogeny of Treeshrews (Mammalia: Scandentia) and the Timescale of Diversification in Southeast Asia. Mol. Phylogenet. Evol. 2011, 60, 358–372. [Google Scholar] [CrossRef]
- Helgen, K.M. Order Scandentia. In Mammal Species of the World, 3rd ed.; Wilson, D.E., Reeder, D.M., Eds.; The Johns Hopkins University Press: Baltimore, MD, USA, 2005; pp. 104–109. [Google Scholar]
- Li, Q.; Ni, X. An Early Oligocene Fossil Demonstrates Treeshrews Are Slowly Evolving “Living Fossils”. Sci. Rep. 2016, 6, 18627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmins, R.J.; Duckworth, J.W.; Robson, C.R.; Walston, J.L. Distribution, Status and Ecology of the Mainland Slender-Tailed Treeshrew Dendrogale murina: Status of Dendrogale murina. Mammal Rev. 2003, 33, 272–283. [Google Scholar] [CrossRef]
- Timmins, R.J. IUCN Red List of Threatened Species: Dendrogale murina. IUCN Red List Threat. Species 2016, e.T41490A22278606. [Google Scholar] [CrossRef]
- Hawkins, M.T.R. Family Tupaiidae (Treeshrews). In Insectivores, Sloths and Colugos; Handbook of the Mammals of the World; Lynx Edicions: Barcelona, Spain, 2018; pp. 242–269. [Google Scholar]
- Francis, C. Field Guide to the Mammals of South-East Asia, 2nd ed.; Bloomsbury Publishing: London, UK, 2019. [Google Scholar]
- Butler, P.M. The Tupaiid Dentition. In Comparative Biology and Evolutionary Relationships of Tree Shrews; Luckett, W.P., Ed.; Springer US: Boston, MA, USA, 1980; pp. 171–204. ISBN 978-1-4684-1053-2. [Google Scholar]
- Steele, D.G. Dental Variability in the Tree Shrews (Tupaiidae). In Craniofacial Biology of Primates: Symposium of the 4th International Congress of Primatology; Zingeser, M.R., Ed.; Karger: Basel, Switzerland, 1973; pp. 154–179. [Google Scholar]
- Luckett, W.P. The Suggested Evolutionary Relationships and Classification of Tree Shrews. In Comparative Biology and Evolutionary Relationships of Tree Shrews; Luckett, W.P., Ed.; Springer US: Boston, MA, USA, 1980; pp. 3–31. ISBN 978-1-4684-1053-2. [Google Scholar]
- Olson, L.E.; Sargis, E.J.; Martin, R.D. Intraordinal Phylogenetics of Treeshrews (Mammalia: Scandentia) Based on Evidence from the Mitochondrial 12S RRNA Gene. Mol. Phylogenet. Evol. 2005, 35, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.E.; Sargis, E.J.; Olson, L.E. Networks, Trees, and Treeshrews: Assessing Support and Identifying Conflict with Multiple Loci and a Problematic Root. Syst. Biol. 2009, 58, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, K.; Csorba, G.; Hughes, A.C.; Jin, L.; Xiao, Y.; Feng, J. Complete Mitochondrial Genomes Reveal Robust Phylogenetic Signals and Evidence of Positive Selection in Horseshoe Bats. BMC Ecol. Evol. 2021, 21, 199. [Google Scholar] [CrossRef] [PubMed]
- Abramson, N.I.; Bodrov, S.Y.; Bondareva, O.V.; Genelt-Yanovskiy, E.A.; Petrova, T.V. A Mitochondrial Genome Phylogeny of Voles and Lemmings (Rodentia: Arvicolinae): Evolutionary and Taxonomic Implications. PLoS ONE 2021, 16, e0248198. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, A.; Veron, G.; Ropiquet, A.; Jansen van Vuuren, B.; Lécu, A.; Goodman, S.M.; Haider, J.; Nguyen, T.T. Evolutionary History of Carnivora (Mammalia, Laurasiatheria) Inferred from Mitochondrial Genomes. PLoS ONE 2021, 16, e0240770. [Google Scholar] [CrossRef]
- Kundu, S.; Pakrashi, A.; Kamalakannan, M.; Singha, D.; Tyagi, K.; Banerjee, D.; Venkatraman, C.; Kumar, V. Complete Mitogenome of the Endangered and Endemic Nicobar Treeshrew (Tupaia Nicobarica) and Comparison with Other Scandentians. Sci. Rep. 2022, 12, 877. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 September 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antipov, D.; Hartwick, N.; Shen, M.; Raiko, M.; Lapidus, A.; Pevzner, P.A. PlasmidSPAdes: Assembling Plasmids from Whole Genome Sequencing Data. Bioinformatics 2016, 32, btw493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de Novo Metazoan Mitochondrial Genome Annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of Nucleotide Composition at Fourfold Degenerate Sites of Animal Mitochondrial Genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [Green Version]
- De Vries, D.; Beck, R.M.D. Total Evidence Tip-Dating Phylogeny of Platyrrhine Primates and 27 Well-Justified Fossil Calibrations for Primate Divergences. Evol. Biol. 2021. [Google Scholar] [CrossRef]
- Wilson Mantilla, G.P.; Chester, S.G.B.; Clemens, W.A.; Moore, J.R.; Sprain, C.J.; Hovatter, B.T.; Mitchell, W.S.; Mans, W.W.; Mundil, R.; Renne, P.R. Earliest Palaeocene Purgatoriids and the Initial Radiation of Stem Primates. R. Soc. Open Sci. 2021, 8, 210050. [Google Scholar] [CrossRef] [PubMed]
- Mein, P.; Ginsburg, L. Les Mammifères Du Gisement Miocène Inférieur de Li Mae Long, Thaïlande: Systématique, Biostratigraphie et Paléoenvironnement. Geodiversitas 1997, 19, 783–844. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.; de Bruijn, M.H.L.; Coulson, A.R.; Eperon, I.C.; Sanger, F.; Young, I.G. Complete Sequence of Bovine Mitochondrial DNA Conserved Features of the Mammalian Mitochondrial Genome. J. Mol. Biol. 1982, 156, 683–717. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Start | Stop | Length | Direction | fcd | scd | Completeness | Absent Fragments, aa |
|---|---|---|---|---|---|---|---|---|
| Dendrogale murina OP006204 | ||||||||
| ND1 | 2754 | 3710 | 957 | forward | ATG | TAG | complete | - |
| ND2 | 3919 | 4962 | 1044 | forward | ATT | TAG | complete | - |
| COX1 | 5344 | 6891 | 1548 | forward | ATG | AGA | complete | - |
| COX2 | 7028 | 7711 | 684 | forward | ATG | TAG | complete | - |
| ATP8 | 7777 | 7980 | 204 | forward | ATG | TAA | complete | - |
| ATP6 | 7938 | 8618 | 681 | forward | ATG | TAA | complete | - |
| COX3 | 8618 | 9403 | 786 | forward | ATG | TAA | complete | - |
| ND3 | 9472 | 9818 | 347 | forward | ATT | TAA* | complete | - |
| ND4L | 9886 | 10,182 | 297 | forward | ATG | TAA | complete | - |
| ND4 | 10,176 | 11,553 | 1378 | forward | ATG | TAA* | complete | - |
| ND5 | 11,751 | 13,562 | 1812 | forward | ATT | TAA | complete | - |
| ND6 | 14,079 | 13,558 | 522 | reverse | ATG | AGA | complete | - |
| CYTB | 14,152 | 15,291 | 1140 | forward | ATG | TAG | complete | - |
| Dendrogale murina OP006205 | ||||||||
| ND1 | 2754 | 3710 | 957 | forward | ATG | TAG | complete | - |
| ND2 | 3919 | 4962 | 1044 | forward | ATT | TAG | complete | - |
| COX1 | 5344 | 6891 | 1548 | forward | ATG | AGA | complete | - |
| COX2 | 7028 | 7711 | 684 | forward | ATG | TAG | complete | - |
| ATP8 | 7777 | 7980 | 204 | forward | ATG | TAA | complete | - |
| ATP6 | 7938 | 8618 | 681 | forward | ATG | TAA | complete | - |
| COX3 | 8618 | 9403 | 786 | forward | ATG | TAA | complete | - |
| ND3 | 9472 | 9818 | 347 | forward | ATT | TAA* | complete | - |
| ND4L | 9886 | 10,182 | 297 | forward | ATG | TAA | complete | - |
| ND4 | 10,176 | 11,553 | 1378 | forward | ATG | TAA* | complete | - |
| ND5 | 11,752 | 13,563 | 1812 | forward | ATT | TAA | complete | - |
| ND6 | 14,080 | 13,559 | 522 | reverse | ATG | AGA | complete | - |
| CYTB | 14,153 | 15,292 | 1140 | forward | ATG | TAG | complete | - |
| Ptilocercus lowii | ||||||||
| ND1 | 2755 | 3711 | 957 | forward | ATG | TAG | partial | 2914–3418; |
| ND2 | 3916 | 4957 | 1042 | forward | ATA | - | partial | 4074–4134; 4321–4957 |
| COX1 | 5348 | 6889 | 1542 | forward | ATG | TAA | partial | 6565–6619; 6751–6836 |
| COX2 | 7032 | 7715 | 684 | forward | ATG | - | partial | 7421–7715 |
| ATP8 | 7783 | 7989 | 207 | forward | - | TAG | partial | 7783–7896 |
| ATP6 | 7944 | 8624 | 681 | forward | ATG | TAA | partial | 8030–8106; 8496–8516 |
| COX3 | 8624 | 9408 | 785 | forward | ATG | TAA* | partial | 8880–8898 |
| ND3 | 9477 | 9823 | 347 | forward | - | TAA* | partial | 9477–9530; 9711–9757 |
| ND4L | 9897 | 10,151 | 255 | forward | ATG | TAA | partial | 10,023–10,043 |
| ND4 | 10,145 | 11,521 | 1377 | forward | ATG | TAA | partial | 10,235–10,477; 10,640–10,658; 11,275–11,459 |
| ND5 | 11,722 | 13,534 | 1813 | forward | ATT | - | partial | 11,791–12,704; 12,805–12,918; 13,031–13,534 |
| ND6 | 14,057 | 13,537 | 521 | reverse | ATG | - | partial | 13,537–13,918; |
| CYTB | 14,131 | 15,270 | 1140 | forward | - | - | partial | 14,131–14,148; 14,869–14,947; 15,203–15,270 |
| Taxon Separated | Mean | 95% HPD |
|---|---|---|
| Ptilocercus lowii | 46.32 | 57.53–34.28 |
| Dendrogale murina | 35.83 | 45.76–26.72 |
| Tupaia belangeri | 22.33 | 27.89–18.03 |
| Tupaia nicobarica | 16.71 | 21.4–12.59 |
| Tupaia minor | 13.71 | 17.67–9.89 |
| Tupaia tana | 9.34 | 12.35–6.54 |
| Tupaia montana / T. splendidula | 7.08 | 9.58–4.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, T.; Bondareva, O.; Bodrov, S.; Abramov, A.; Abramson, N. The Complete Mitochondrial Genome of Dendrogale murina (Tupaiidae) and Phylogeny of Scandentia. Genes 2023, 14, 624. https://doi.org/10.3390/genes14030624
Petrova T, Bondareva O, Bodrov S, Abramov A, Abramson N. The Complete Mitochondrial Genome of Dendrogale murina (Tupaiidae) and Phylogeny of Scandentia. Genes. 2023; 14(3):624. https://doi.org/10.3390/genes14030624
Chicago/Turabian StylePetrova, Tatyana, Olga Bondareva, Semyon Bodrov, Alexei Abramov, and Natalia Abramson. 2023. "The Complete Mitochondrial Genome of Dendrogale murina (Tupaiidae) and Phylogeny of Scandentia" Genes 14, no. 3: 624. https://doi.org/10.3390/genes14030624
APA StylePetrova, T., Bondareva, O., Bodrov, S., Abramov, A., & Abramson, N. (2023). The Complete Mitochondrial Genome of Dendrogale murina (Tupaiidae) and Phylogeny of Scandentia. Genes, 14(3), 624. https://doi.org/10.3390/genes14030624