
The Expanding Phenotypical Spectrum of WARS2-Related Disorder: Four Novel Cases with a Common Recurrent Variant

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Variants and Additional Patients
2.2. Collection of Biosamples
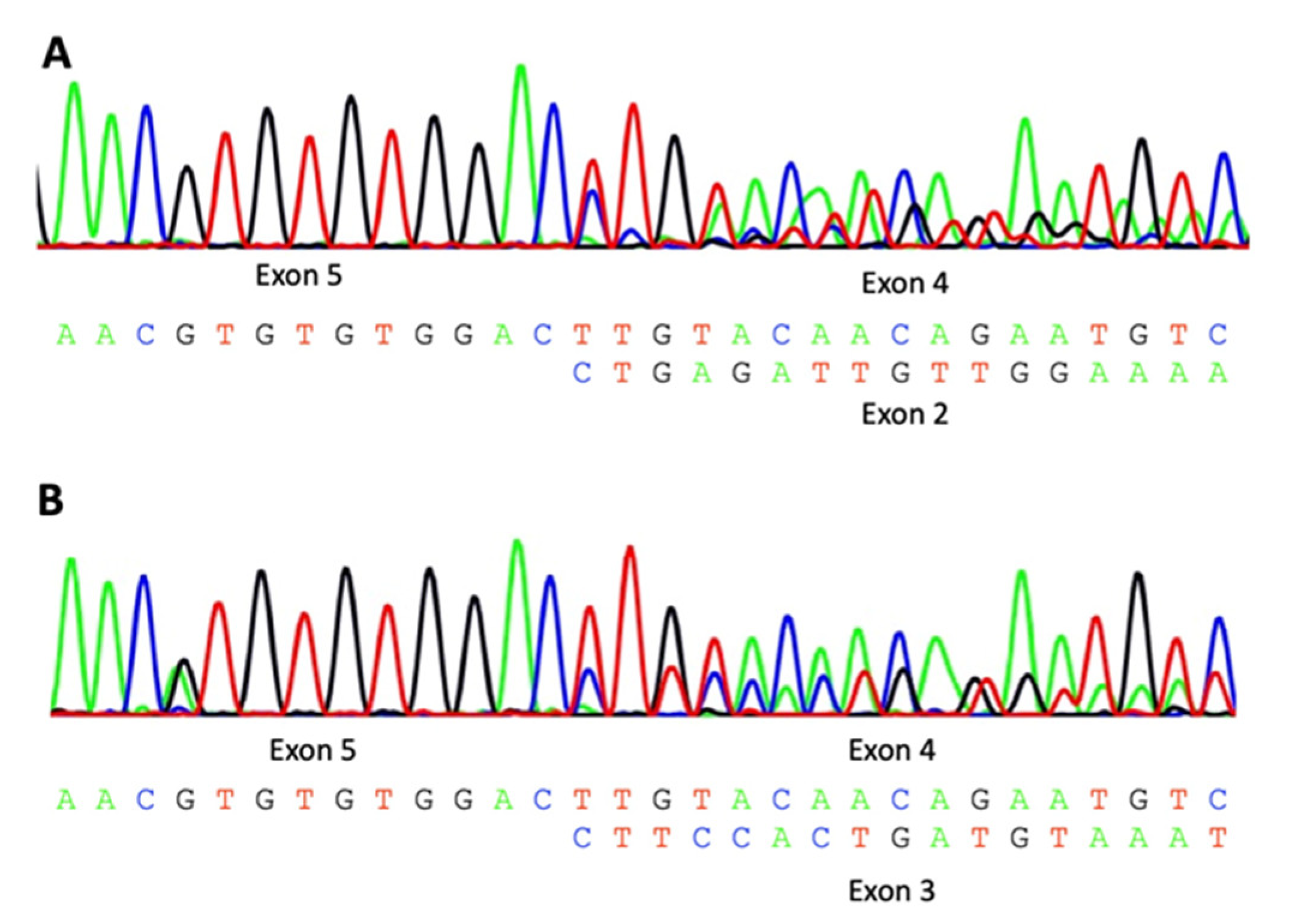
2.3. Sanger Sequencing and Sequencing of cDNA
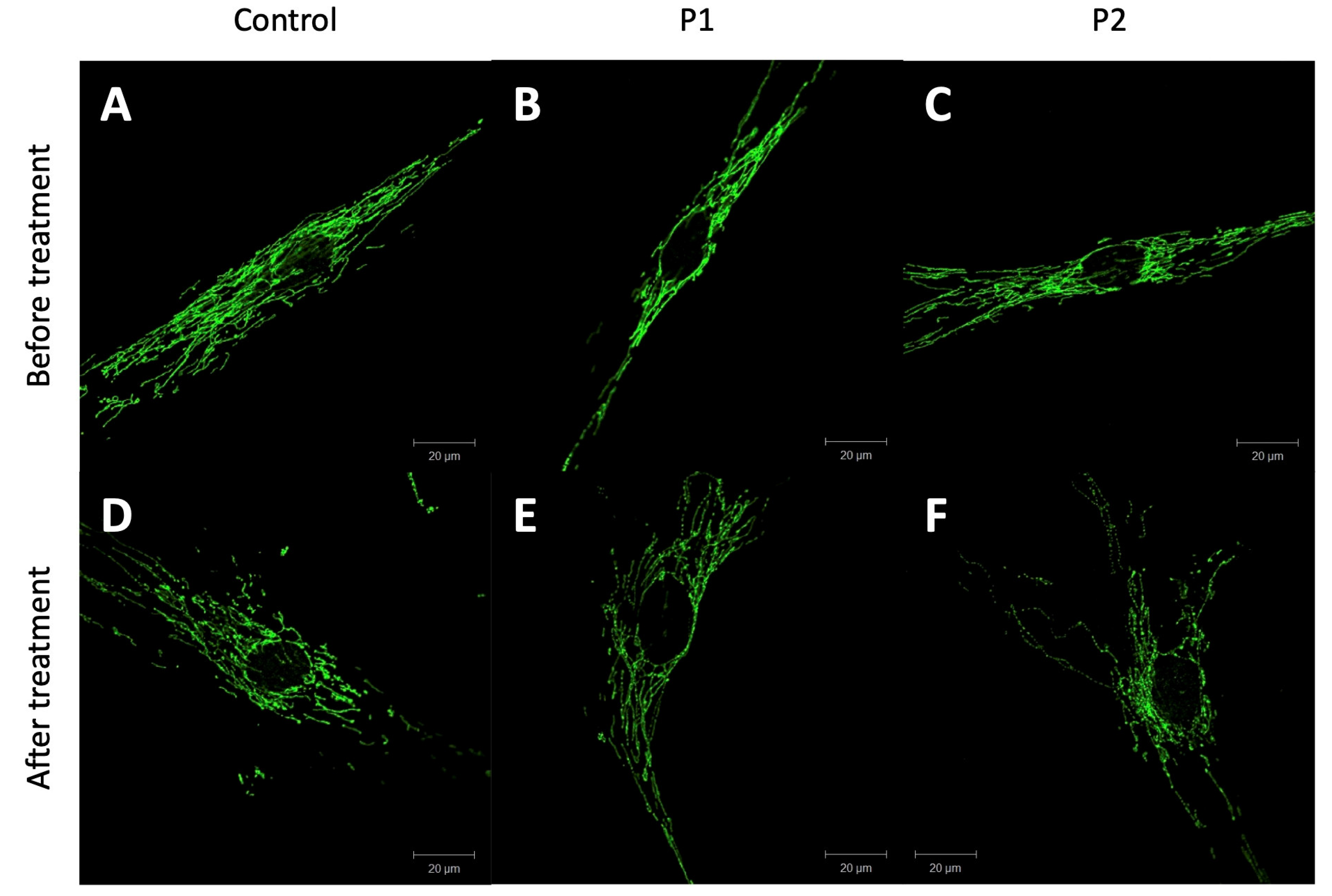
2.4. Staining of the Mitochondrial Network
2.5. Western Blotting of MT-CO2
3. Results
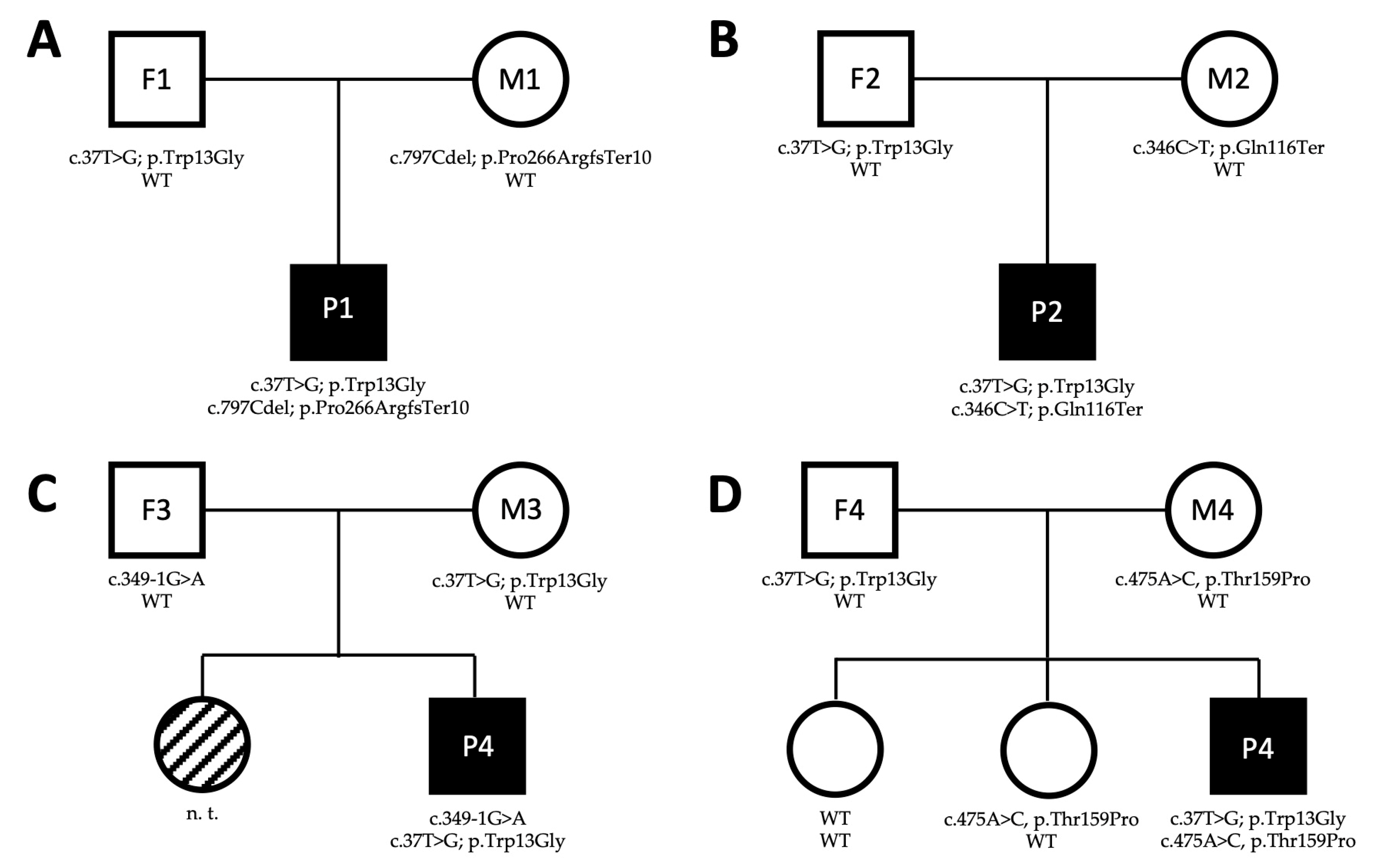
3.1. Case Reports
3.1.1. Patient 1
3.1.2. Patient 2
3.1.3. Patient 3
3.1.4. Patient 4
3.2. Characterization of WARS2 Variants
3.3. Mitochondrial Integrity
3.4. Respiratory Chain and WARS2 Level
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martinez-Dominguez, M.T.; Justesen, J.; Kruse, T.A.; Hansen, L.L. Assignment of the Human Mitochondrial Tryptophanyl-TRNA Synthetase (WARS2) to 1p13.3-->p13.1 by Radiation Hybrid Mapping. Cytogenet. Cell Genet. 1998, 83, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Ognjenović, J.; Simonović, M. Human Aminoacyl-TRNA Synthetases in Diseases of the Nervous System. RNA Biol. 2018, 15, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musante, L.; Püttmann, L.; Kahrizi, K.; Garshasbi, M.; Hu, H.; Stehr, H.; Lipkowitz, B.; Otto, S.; Jensen, L.R.; Tzschach, A.; et al. Mutations of the Aminoacyl-TRNA-Synthetases SARS and WARS2 Are Implicated in the Etiology of Autosomal Recessive Intellectual Disability: MUSANTE ET AL. Hum. Mutat. 2017, 38, 621–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theisen, B.E.; Rumyantseva, A.; Cohen, J.S.; Alcaraz, W.A.; Shinde, D.N.; Tang, S.; Srivastava, S.; Pevsner, J.; Trifunovic, A.; Fatemi, A. Deficiency of WARS2, Encoding Mitochondrial Tryptophanyl TRNA Synthetase, Causes Severe Infantile Onset Leukoencephalopathy. Am. J. Med. Genet. Part A 2017, 173, 2505–2510. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Timal, S.; Venselaar, H.; Wintjes, L.T.; Kopajtich, R.; Feichtinger, R.G.; Onnekink, C.; Mühlmeister, M.; Brandt, U.; Smeitink, J.A.; et al. Biallelic Variants in WARS2 Encoding Mitochondrial Tryptophanyl-TRNA Synthase in Six Individuals with Mitochondrial Encephalopathy. Hum. Mutat. 2017, 38, 1786–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, E.A.; Frucht, S.J.; Thompson, K.; Wolfe, L.A.; Yokoyama, T.; Bertoni, M.; Huang, Y.; Sincan, M.; Adams, D.R.; Taylor, R.W.; et al. Biallelic Mutations in Mitochondrial Tryptophanyl-TRNA Synthetase Cause Levodopa-Responsive Infantile-Onset Parkinsonism. Clin. Genet. 2018, 93, 712–718. [Google Scholar] [CrossRef] [Green Version]
- Vantroys, E.; Smet, J.; Vanlander, A.V.; Vergult, S.; De Bruyne, R.; Roels, F.; Stepman, H.; Roeyers, H.; Menten, B.; Van Coster, R. Severe Hepatopathy and Neurological Deterioration after Start of Valproate Treatment in a 6-Year-Old Child with Mitochondrial Tryptophanyl-TRNA Synthetase Deficiency. Orphanet. J. Rare Dis. 2018, 13, 80. [Google Scholar] [CrossRef]
- Maffezzini, C.; Laine, I.; Dallabona, C.; Clemente, P.; Calvo-Garrido, J.; Wibom, R.; Naess, K.; Barbaro, M.; Falk, A.; Donnini, C.; et al. Mutations in the Mitochondrial Tryptophanyl-tRNA Synthetase Cause Growth Retardation and Progressive Leukoencephalopathy. Mol. Genet. Genomic. Med. 2019, 7, e654. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, C.; Silva, L.; Pereira, C.; Vieira, L.; Leão Teles, E.; Rodrigues, E.; Campos, T.; Janeiro, P.; Gaspar, A.; Dupont, J.; et al. Targeted next Generation Sequencing Identifies Novel Pathogenic Variants and Provides Molecular Diagnoses in a Cohort of Pediatric and Adult Patients with Unexplained Mitochondrial Dysfunction. Mitochondrion 2019, 47, 309–317. [Google Scholar] [CrossRef]
- Virdee, M.; Swarnalingam, E.; Kozenko, M.; Tarnopolsky, M.; Jones, K. Expanding the Phenotype: Neurodevelopmental Disorder, Mitochondrial, With Abnormal Movements and Lactic Acidosis, With or Without Seizures (NEMMLAS) Due to WARS2 Biallelic Variants, Encoding Mitochondrial Tryptophanyl-TRNA Synthase. J. Child Neurol. 2019, 34, 778–781. [Google Scholar] [CrossRef]
- Hübers, A.; Huppertz, H.; Wortmann, S.B.; Kassubek, J. Mutation of the WARS2 Gene as the Cause of a Severe Hyperkinetic Movement Disorder. Mov. Disord. Clin. Pract. 2020, 7, 88–90. [Google Scholar] [CrossRef]
- Martinelli, S.; Cordeddu, V.; Galosi, S.; Lanzo, A.; Palma, E.; Pannone, L.; Ciolfi, A.; Di Nottia, M.; Rizza, T.; Bocchinfuso, G.; et al. Co-Occurring WARS2 and CHRNA6 Mutations in a Child with a Severe Form of Infantile Parkinsonism. Park. Relat. Disord. 2020, 72, 75–79. [Google Scholar] [CrossRef]
- Ilinca, A.; Kafantari, E.; Puschmann, A. A Relatively Common Hypomorphic Variant in WARS2 Causes Monogenic Disease. Park. Relat. Disord. 2022, 94, 129–131. [Google Scholar] [CrossRef]
- Skorvanek, M.; Rektorova, I.; Mandemakers, W.; Wagner, M.; Steinfeld, R.; Orec, L.; Han, V.; Pavelekova, P.; Lackova, A.; Kulcsarova, K.; et al. WARS2 Mutations Cause Dopa-Responsive Early-Onset Parkinsonism and Progressive Myoclonus Ataxia. Park. Relat. Disord. 2022, 94, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Zurek, B.; Ellwanger, K.; Vissers, L.E.L.M.; Schüle, R.; Synofzik, M.; Töpf, A.; de Voer, R.M.; Laurie, S.; Matalonga, L.; Gilissen, C.; et al. Solve-RD: Systematic Pan-European Data Sharing and Collaborative Analysis to Solve Rare Diseases. Eur. J. Hum. Genet. 2021, 29, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Matalonga, L.; Hernández-Ferrer, C.; Piscia, D.; Cohen, E.; Solve-RD SNV-indel Working Group; Cuesta, I.; Danis, D.; Denommé-Pichon, A.-S.; Duffourd, Y.; Gilissen, C.; et al. Solving Patients with Rare Diseases through Programmatic Reanalysis of Genome-Phenome Data. Eur. J. Hum. Genet. 2021, 29, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Lochmüller, H.; Badowska, D.M.; Thompson, R.; Knoers, N.V.; Aartsma-Rus, A.; Gut, I.; Wood, L.; Harmuth, T.; Durudas, A.; Graessner, H.; et al. RD-Connect, NeurOmics and EURenOmics: Collaborative European Initiative for Rare Diseases. Eur. J. Hum. Genet. 2018, 26, 778–785. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Diaw, S.H.; Ganos, C.; Zittel, S.; Plötze-Martin, K.; Kulikovskaja, L.; Vos, M.; Westenberger, A.; Rakovic, A.; Lohmann, K.; Dulovic-Mahlow, M. Mutant WDR45 Leads to Altered Ferritinophagy and Ferroptosis in β-Propeller Protein-Associated Neurodegeneration. IJMS 2022, 23, 9524. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Voges, L.; Rakovic, A.; Kasten, M.; Vandebona, H.; Hemmelmann, C.; Lohmann, K.; Orolicki, S.; Ramirez, A.; Schapira, A.H.V.; et al. Mutant Parkin Impairs Mitochondrial Function and Morphology in Human Fibroblasts. PLoS ONE 2010, 5, e12962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | P1 | P2 | P3 | P4 | Literature (n = 24) 1 [3,4,5,6,7,8,9,10,11,12,13,14] |
|---|---|---|---|---|---|
| Sex | Male | Male | Male | Male | Male 11/24 (45.8%) |
| Age of onset | 0.5 y | 9 y | 13 y | 1 y | Mean 2.0 y [range 0–12 y] |
| Age at last examination | 20 y | 20 y | 38 y | 2 y | Mean 16.4 y [range 0–52 y] |
| First signs and symptoms | Tremor | Tremor, intellectual disability | Tremor | Tremor, developmental delay | Hyperkinetic MD 8/22 |
| Developmental delay 8/22 | |||||
| Others 2 6/22 | |||||
| Tremor | yes | yes | yes | yes | 9/9 |
| Parkinsonism | yes | yes | yes | yes | 5/5 |
| Dystonia | yes | yes | yes | no | 12/12 |
| Other hyperkinetic MD 3 | no | Myoclonic jerks | Myoclonic jerks | no | 6/6 |
| Ataxia | no | no | no | yes | 8/8 |
| Increased muscle tone | yes | yes | yes | yes | 10/10 |
| (Axial) hypotonia | no | no | no | yes | 8/8 |
| Muscular weakness | no | no | no | no | 4/4 |
| Seizures | no | no | no | no | 7/10 |
| Developmental delay | no | no | no | yes | 7/7 |
| Intellectual disorder | mild | mild | mild | (mild to moderate) 4 | 16/20 |
| Mild to moderate | 6/15 | ||||
| severe | 9/15 | ||||
| Lactate acidosis | no | no | no | no | 3/4 |
| L-Dopa response | good | good | good | good | 6/8 |
| MRI abnormal | no | Pallidal T2 hypointensity | no | no | 5/10 5 |
| DaTScan abnormal | n. a. | Yes | yes | n. a. | 3/3 |
| Mutation | Patients | Families | |||
|---|---|---|---|---|---|
| c.622G>T | p.Glu208Ter | 1 | 3.6% | 1 | 4.3% |
| c.1054G>A | p.Glu352Lys | 1 | 3.6% | 1 | 4.3% |
| c.134G>T | p.Gly45Val | 1 | 3.6% | 1 | 4.3% |
| c.231C>G | p.His77Gln | 1 | 3.6% | 1 | 4.3% |
| c.487C>T | p.Leu163Phe | 1 | 3.6% | 1 | 4.3% |
| c.679A>G | p.Met227Val | 1 | 3.6% | 1 | 4.3% |
| c.683C>G | p.Ser228Trp | 1 | 3.6% | 1 | 4.3% |
| c.532G>C | p.Val178Leu | 1 # | 3.6% | 1 | 4.3% |
| c.1045G>A | p.Val349Leu | 1 | 3.6% | 1 | 4.3% |
| c.346C>T | p.Gln116Ter | 1 | 3.6% | 1 | 4.3% |
| c.349-1G>A | Splice alteration | 1 | 3.6% | 1 | 4.3% |
| c.475A>C | p.Thr159Pro | 1 | 3.6% | 1 | 4.3% |
| c.149G>A | p.Gly50Asp | 2 | 7.1% | 2 | 8.7% |
| c.325delA | p.Ser109Alafs*159 | 2 | 7.1% | 1 | 4.3% |
| c.298_300delCTT | p.Leu100del | 3 | 10.7% | 2 | 8.7% |
| c.91-8725_348+27116del36099 | p.Lys31_Gln116del | 3 | 10.7% | 2 | 8.7% |
| c.833T>G | p.Val278Gly | 3 | 10.7% | 2 | 8.7% |
| c.797delC | p.Pro266Argfs*10 | 5 | 17.9% | 3 | 13.0% |
| c.938A>T | p.Lys313Met | 9 | 32.1% | 7 | 30.4% |
| c.37T>G | p.Trp13Gly | 16 | 57.1% | 9 | 39.1% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauly, M.G.; Korenke, G.C.; Diaw, S.H.; Grözinger, A.; Cazurro-Gutiérrez, A.; Pérez-Dueñas, B.; González, V.; Macaya, A.; Serrano Antón, A.T.; Peterlin, B.; et al. The Expanding Phenotypical Spectrum of WARS2-Related Disorder: Four Novel Cases with a Common Recurrent Variant. Genes 2023, 14, 822. https://doi.org/10.3390/genes14040822
Pauly MG, Korenke GC, Diaw SH, Grözinger A, Cazurro-Gutiérrez A, Pérez-Dueñas B, González V, Macaya A, Serrano Antón AT, Peterlin B, et al. The Expanding Phenotypical Spectrum of WARS2-Related Disorder: Four Novel Cases with a Common Recurrent Variant. Genes. 2023; 14(4):822. https://doi.org/10.3390/genes14040822
Chicago/Turabian StylePauly, Martje G., G. Christoph Korenke, Sokhna Haissatou Diaw, Anne Grözinger, Ana Cazurro-Gutiérrez, Belén Pérez-Dueñas, Victoria González, Alfons Macaya, Ana Teresa Serrano Antón, Borut Peterlin, and et al. 2023. "The Expanding Phenotypical Spectrum of WARS2-Related Disorder: Four Novel Cases with a Common Recurrent Variant" Genes 14, no. 4: 822. https://doi.org/10.3390/genes14040822
APA StylePauly, M. G., Korenke, G. C., Diaw, S. H., Grözinger, A., Cazurro-Gutiérrez, A., Pérez-Dueñas, B., González, V., Macaya, A., Serrano Antón, A. T., Peterlin, B., Božović, I. B., Maver, A., Münchau, A., & Lohmann, K. (2023). The Expanding Phenotypical Spectrum of WARS2-Related Disorder: Four Novel Cases with a Common Recurrent Variant. Genes, 14(4), 822. https://doi.org/10.3390/genes14040822