
Rare Coding Variants in Patients with Non-Syndromic Vestibular Dysfunction

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approvals and Subject Recruitment
- Cohort 1: For the pilot study, five children who were diagnosed with vestibular dysfunction and had a family history of vertigo were recruited into the study (Table 1). Patients were excluded if there was a diagnosis of hearing loss, known neurologic disorders, or other significant comorbidities. The five enrolled patients did not have any history of head trauma, notable past medical, prenatal, and neonatal events, and inner ear abnormalities seen by 2D high-resolution temporal bone CT or MRI. The results of audiometric testing and tympanometry were within the normal limits. Saliva samples were obtained from the study participants using Oragene saliva kits (DNA Genotek, Ottawa, ON, Canada). Genomic DNA was isolated from saliva using the manufacturer’s protocol.

{kind=link}
{kind=link}
| ID 1 | Age at Initial Consult | Sex | Ethnicity | Family History of Migraine | Duration of Symptoms | Clinical Diagnosis |
|---|---|---|---|---|---|---|
| 1 | 6 years | F | White non-Hispanic | No | 2 years | Benign paroxysmal vertigo of childhood |
| 2 | 11 years | M | Hispanic | No | 5 months | Vertigo |
| 3 | 13 years | F | White non-Hispanic | Yes | 9 years | Vestibular migraine |
| 4 | 9 years | F | White non-Hispanic | Yes | 6 years | Vestibular migraine |
| 5 2 | 12 years | F | White non-Hispanic | Yes | 8 years | Left-sided vestibular weakness |
- Cohort 2: Meniere’s disease patients were recruited and diagnosed from different Spanish hospitals following the diagnostic criteria defined by the International Classification Committee for Vestibular Disorders of the Barany Society [9]. Other vestibular diseases were excluded after performing a complete audio-vestibular assessment, including MRI. The DNA from these patients were collected and extracted from blood or saliva using the QIAamp DNA Mini Kit (Qiagen, Venlo, the Netherlands) and Oragene saliva kits, respectively.
- Cohort 3: European–American families with AIS were enrolled as previously described [10,11,12]. The families were selected for sequencing if the proband had no co-occurring congenital deformity or genetic disorder and was diagnosed with AIS based on a standing anteroposterior spinal radiograph showing ≥10° curvature by the Cobb method with pedicle rotation. In total, 3 to 4 AIS-affected individuals from each of the 28 families were selected for exome sequencing based on the scoliotic curve severity, the availability of DNA samples, and the genetic distance across the individuals [10,11].
- Cohort 4: A second cohort of individuals with AIS (Table S1) were enrolled as above with added consent to obtain the morphological measurements of the semicircular canals using the three-dimensional reconstruction of images from T2-weighted MRI [13]. This previous study was performed to examine the association between the occurrence of semicircular canal defects and spine deformity. From cohort 4, eleven individuals with differences in angulation between the axial plane and the lateral semicircular canal when comparing the right and left inner ears (from here on referred to as LSCC asymmetry) were selected for Sanger sequencing of exons within two genes: HMX3 and OTOP1. Of note, out of eleven selected individuals with subtle changes in their inner ear structure, ten were previously diagnosed with AIS (Table S1), while the eleventh individual was designated as an AIS control due to normal spine curvature.
- Cohort 5: Two American patients with vestibular schwannomas who underwent tumor resection via a translabyrinthine approach provided vestibular tissue samples that would otherwise be discarded during surgery. One patient self-identified as White/Caucasian, while the second patient refused to disclose their ethnicity. Both patients did not experience any preoperative symptoms of vertigo, suggesting normal vestibular function despite the ipsilateral nerve sheath tumor. Immediately upon removal, the vestibular tissue samples were placed in RNAlater (Invitrogen/Thermo Fisher, Waltham, MA, USA). The samples were stored at 4 °C and processed within 30 days.
2.2. Exome and Sanger Sequencing of DNA Samples
2.3. Variant Prioritization and Gene Network Analysis
| ID | Gene | RefSeq NM_# | Variant | Highest MAF in Databases 1 | Damaging Prediction | Scaled CADD Score | Known Literature on Gene [References] | MIM# |
|---|---|---|---|---|---|---|---|---|
| 1 | TMEM130 | 001134450 | c.52C>T (p.(Leu18Phe)) | 0 | SI, MC | 24.1 | High expression in developing mouse utricular hair cells [18] | n.a. |
| 1 | LRP1 2 | 002332 | c.13471G>C (p.(Asp4491His)) | (gA)OTH: 9.5 × 10–4 | ppHD, ppHV, LRT, MT | 24.8 | Associated with migraine [19] | 107770 |
| 1 | ECM1 2 | 001202858 | c.1126T>C (p.(Cys376Arg)) | (AU)Lat: 9.9 × 10–5 | SI, ppHD, ppHV, LRT, MT, FA, PR, mSVM, mLR, MC | 25.5 | Associated with migraine [20,21] | 602201 |
| 1 | CARF 2 | 001352679 | c.1718C>T (p.(Ser573Phe)) | (AU)OTH: 1.1 × 10–4 | SI, ppHD, ppHV, MT | 27.9 | Associated with migraine [20,22] | 607586 |
| 2 | CTIF | 014772 | c.1547A>G (p.(Gln516Arg)) | (AU)Eur: 3.95 × 10–4 | LRT, fMKL | 20.3 | Associated with migraine [19] | 613178 |
| 2 | KIF6 | 001289024 | c.1838G>A (p.(Arg613Gln)) | (AU)Eur: 7.3 × 10–5 | ppHD, MT, fMKL | 23.6 | High expression in developing mouse utricular hair cells [18] | 613919 |
| 2 | KLC2 2 | 001134774 | c.976C>T (p.(Gln326*)) | 0 | LRT, fMKL | 38.0 | Knockdown in zebrafish resulted in twisted tail and inability to swim [23] | 611729 |
| 2 | OTOP2 | 178160 | c.379A>G (p.(Ser127Gly)) | (gA)NFE: 1.5 × 10–5 | MT, fMKL | 22.0 | Maps to a candidate region of USH1G [24] | 607827 |
| 3 | HMX3 | 001105574 | c.534G>T (p.(Glu178Asp)) | Bravo: 1.6 × 10–5 | ppHD, LRT, MT, FA, mLR, MC | 17.8 | KO mice exhibit abnormal circling behavior and vestibular defects [25] | 613380 |
| 3 | LRP1 | 002332 | c.13169A>C (p.(Asp4390Thr)) | 0 | fMKL | 22.0 | Associated with migraine [19] | 107770 |
| 4 | MYO5A 2 | 000259 | c.3257G>A (p.(Ser1086Asn)) | 0 | LRT, fMKL | 20.6 | Associated with Elejalde syndrome; involved in neurologic development [26] | 276903 |
| 4 | MYO5A 2 | 000259 | c.343C>A (p.(Gln115Lys)) | 0 | SI, LRT, MT, FA, mLR, MC, fMKL | 23.5 | ||
| 4 | RNF213 | 001256071 | c.9197C>T (p.(Pro3066Leu)) | (AU)Eur: 4.2 × 10–5 | SI, LRT, MT, PR, fMKL | 25.3 | Associated with migraine [22] | 613768 |
| 4 | APOE | 000041 | c.875G>T (p.(Arg292Leu)) | (AU)Eur: 2.1 × 10–5 | SI, ppHD, ppHV, PR, MC, fMKL | 31.0 | Associated with migraine [27,28] | 107741 |
| 5 | LAMA2 2 | 000426 | c.6229G>A (p.(Ala2077Thr)) | (gA)NFE: 4.6 × 10–4 | MT, fMKL | 16.9 | Abnormal cochlear and vestibular morphology in homozygous dy/dy mice [29] | 156225 |
| 5 | SLC35D2 | 007001 | c.601A>G (p.(Lys201Glu)) | (gA)NFE: 1.5 × 10–5 | SI, LRT, MT, PR, MC, fMKL | 29.1 | Associated with migraine [19] | 609182 |
| 5 | OTOP1 | 177998 | c.164A>G (p.(Gln55Arg)) | (AU)Eur: 1.98 × 10–4 | MT, MC | 17.3 | Associated with non-syndromic vestibular disorder with otoconial agenesis in mice [30] | 607806 |
2.4. RNA Isolation and RT-PCR for Human Vestibular Tissues
3. Results
3.1. Rare Genetic Variants in Pediatric Patients with Early Onset Vestibular Dysfunction
- Patient 1: For two years prior to consult, a six-year-old female experienced intermittent vertigo lasting 15–60 min, occurring 2–3 times per month, and without associated tinnitus, hearing loss, headaches, and visual changes. She had abnormal bilateral findings with the head impulse test and was diagnosed with benign paroxysmal vertigo of childhood (BPVC). Exome sequencing revealed that she had three rare variants in migraine genes, LRP1 c.13471G>C (p.(Asp4491His)), ECM1 c.1126T>C (p.(Cys376Arg)), and CARF c.1718C>T (p.(Ser573Phe)) [19,20,21,22]. At the time of consult, patient 1 did not exhibit migraine symptoms; however, BPVC is considered a migraine precursor [35]. An additional variant in a gene highly expressed in developing mouse utricular hair cell [18], TMEM130 c.52C>T (p.(Leu18Phe)), was also found.
- Patient 2: An 11-year-old male reported multiple episodes of vertigo lasting minutes to hours during the last five months prior to consultation. He did not have tinnitus, hearing loss, or headaches. His head impulse test had abnormal findings bilaterally, and caloric testing revealed a 21% reduced response on the right side. Rare variants were noted in genes previously associated with neurodevelopmental disorders [23,36]: KIF6 c.1838G>A (p.(Arg613Gln)) [36] and KLC2 c.976C>T (p.(Gln326*)). In addition, he had a rare variant in another migraine-associated gene, CTIF c.1547A>G (p.(Gln516Arg)) [19]. A fourth rare variant, c.379A>G (p.(Ser127Gly)), was found within OTOP2, a gene which maps to the candidate region of Usher syndrome 1G (USH1G) [24], which includes congenital hearing loss, vestibular areflexia, and adolescent-onset retinitis pigmentosa. Note that patient 2 does not have Usher syndrome, hearing loss, or eye disease.
- Patient 3: A 13-year-old female presented with on-and-off vertigo in the previous nine years and episodes of migraine headaches separate from the vertigo attacks. The clinical evaluation revealed an abnormal right-sided head impulse test. Based on her presentation, she was diagnosed with probable vestibular migraine. A damaging variant in the homeobox family protein HMX3 c.534G>T (p.(Glu178Asp)) was identified from her exome sequence data. The knockout of HMX3 in mice led to abnormal circling behavior and severe structural defects in the inner ear vestibule [25]; however, prior to this work, there was no strong evidence of vertigo-causal HMX3 variants in humans [37]. In addition, a rare variant, c.13169A>C (p.(Asn4390Thr)), was identified within the migraine-associated gene LRP1 and was deemed pathogenic by the bioinformatic predictor fathmmMKL.
- Patient 4: A nine-year-old female had recurrent, severe headaches which were unresponsive to a drug cocktail containing Naproxen, Prochlorperazine, and Diphenhydramine. Her headaches were associated with photophobia, phonophobia, vertigo, nausea, vomiting, and occasional tinnitus. The clinical evaluation revealed bilateral torsional and rotational gaze nystagmus, and an abnormal head impulse test on the right. Based on her presentation, she was diagnosed with vestibular migraine. She has novel rare variants in two migraine-associated genes [22,27,28,38]: APOE c.875G>T (p.(Arg292Leu)) [26,33] and RNF213 c.9197C>T (p.(Pro3066Leu)). Additionally, two variants within MYO5A c.3257G>A (p.(Ser1086Asn)) and c.343C>A (p.(Gln115Lys)), a gene previously implicated in neurodevelopment [26], were identified from her exome sequence data. The Sanger sequencing of maternal DNA revealed that these two MYO5A variants were inherited in cis.
- Patient 5: A 12-year-old female presented with an 8-year history of episodic vertigo, lasting minutes to hours, and occasionally occurring when performing gymnastics. The clinical evaluation revealed a bilaterally abnormal head impulse test and 50% left canal paresis on caloric testing. She was diagnosed with left-sided vestibular weakness. A rare missense variant c.6229G>A (p.(Ala2077Thr)) was identified in LAMA2, which is known for merosin-deficient congenital muscular dystrophy 1A (MDC1A; MIM 607855) [29]. Furthermore, Lama2-knockout mice demonstrated cochlear and vestibular malformations [29]. Additionally, two rare variants—c.601A>G (p.(Lys201Glu)) in the migraine-associated gene SLC35D2 [19] and c.164A>G (p.(Gln55Arg)) within OTOP1—were identified in the exome data of patient 5. The knockout of Otop1 previously resulted in non-syndromic vestibular defects and otoconial agenesis in mice [30].
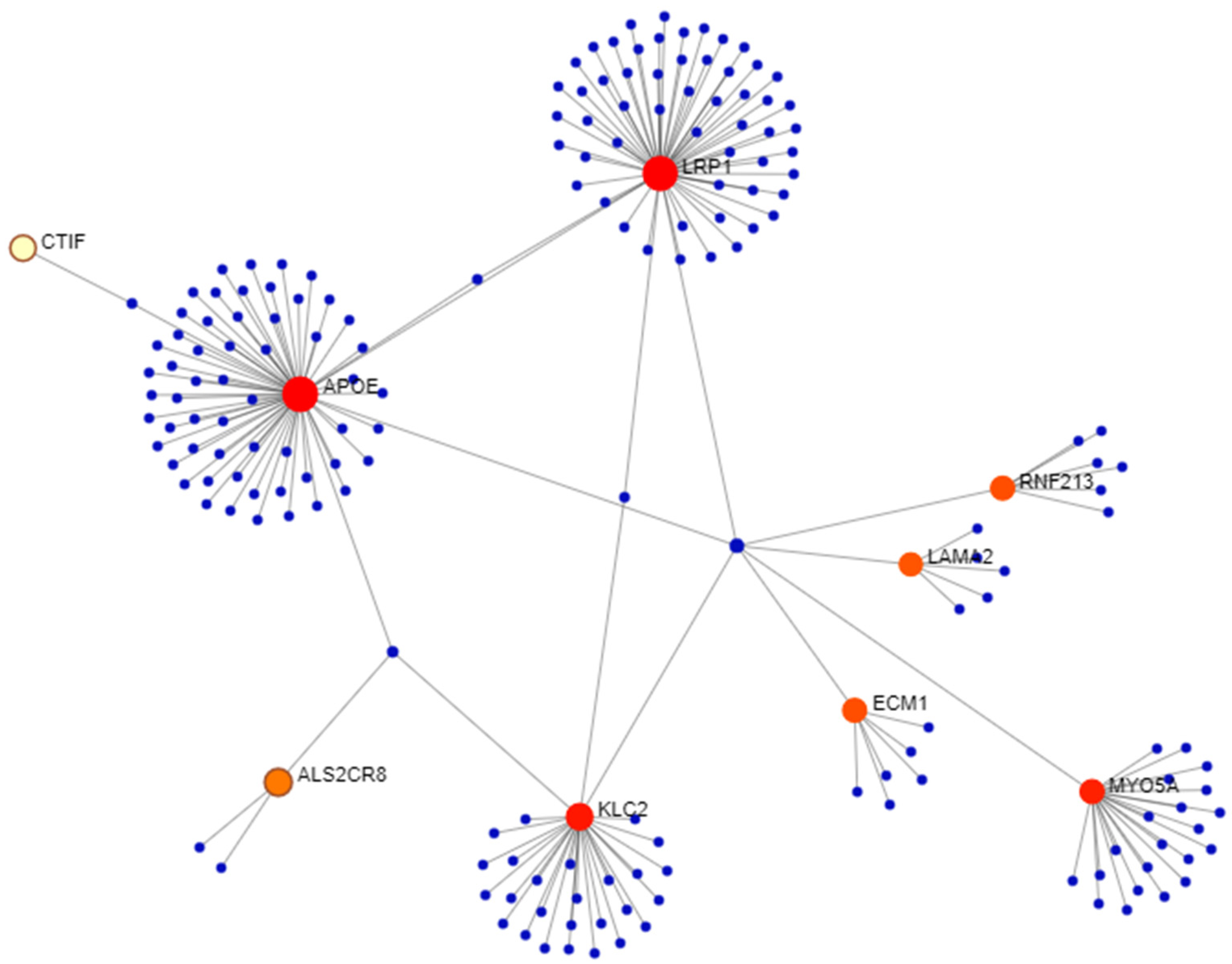
3.2. Network Analysis
3.3. Rare Genetic Variants in Spanish Patients with Meniere’s Disease
3.4. Genetic Variants in European–American Probands with AIS and LSCC Asymmetry
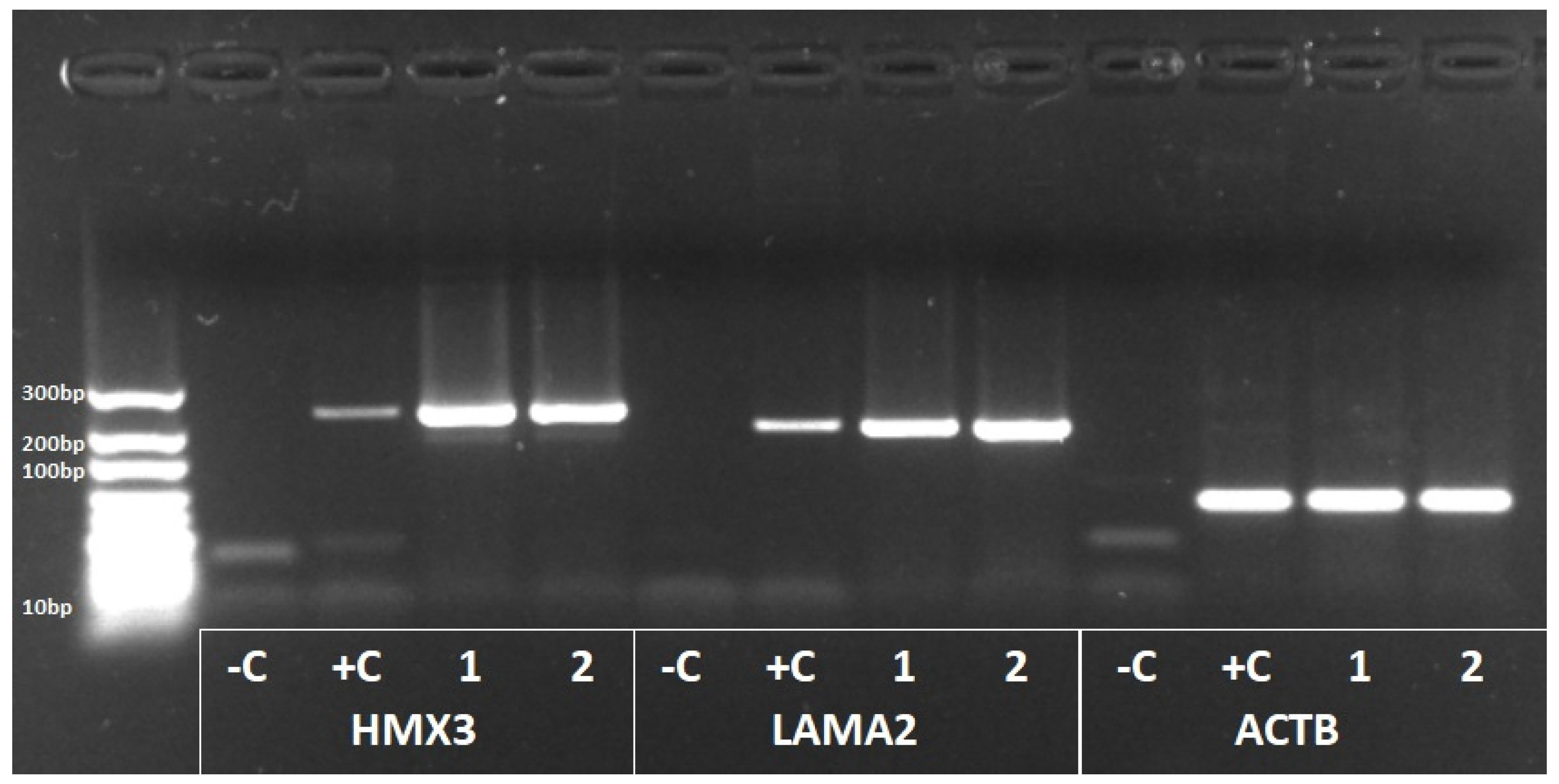
3.5. HMX3 and LAMA2 Expression in Human Vestibular Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yardley, L.; Owen, N.; Nazareth, I.; Luxon, L. Prevalence and presentation of dizziness in a general practice community sample of working age people. Br. J. Gen. Pract. 1998, 48, 1131–1135. [Google Scholar] [PubMed]
- Frejo, L.; Giegling, I.; Teggi, R.; Lopez-Escamez, J.A.; Rujescu, D. Genetics of vestibular disorders: Pathophysiological insights. J. Neurol. 2016, 263 (Suppl. 1), S45–S53. [Google Scholar] [PubMed]
- Hawasli, A.H.; Hullar, T.E.; Dorward, I.G. Idiopathic scoliosis and the vestibular system. Eur. Spine J. 2015, 24, 227–233. [Google Scholar]
- Requena, T.; Espinosa-Sanchez, J.M.; Cabrera, S.; Trinidad, G.; Soto-Varela, A.; Santos-Perez, S.; Teggi, R.; Perez, P.; Batuecas-Caletrio, A.; Fraile, J.; et al. Familial clustering and genetic heterogeneity in Meniere’s disease. Clin. Genet. 2014, 85, 245–252. [Google Scholar] [PubMed]
- Paz-Tamayo, A.; Perez-Carpena, P.; Lopez-Escamez, J.A. Systematic review of prevalence studies and familial aggregation in vestibular migraine. Front. Genet. 2020, 11, 954. [Google Scholar]
- Fancello, V.; Palma, S.; Monzani, D.; Pelucchi, S.; Genovese, E.; Ciorba, A. Vertigo and dizziness in children: An update. Children 2021, 8, 1025. [Google Scholar]
- Richter, G.M.; Wagner, G.; Reichenmiller, K.; Staufenbiel, I.; Martins, O.; Löscher, B.S.; Holtgrewe, M.; Jepsen, S.; Dommisch, H.; Schaefer, A.S. Exome Sequencing of 5 Families with Severe Early-Onset Periodontitis. J. Dent. Res. 2022, 101, 151–157. [Google Scholar]
- Krenn, M.; Wagner, M.; Hotzy, C.; Graf, E.; Weber, S.; Brunet, T.; Lorenz-Depiereux, B.; Kasprian, G.; Aull-Watschinger, S.; Pataraia, E.; et al. Diagnostic exome sequencing in non-acquired focal epilepsies highlights a major role of GATOR1 complex genes. J. Med. Genet. 2020, 57, 624–633. [Google Scholar]
- Lopez-Escamez, J.A.; Carey, J.; Chung, W.H.; Goebel, J.A.; Magnusson, M.; Mandalà, M.; Newman-Toker, D.E.; Strupp, M.; Suzuki, M.; Trabalzini, F.; et al. Diagnostic criteria for Menière’s disease. J. Vestib. Res. 2015, 25, 1–7. [Google Scholar]
- Baschal, E.E.; Wethey, C.I.; Swindle, K.; Baschal, R.M.; Gowan, K.; Tang, N.L.S.; Alvarado, D.M.; Haller, G.E.; Dobbs, M.B.; Taylor, M.R.G.; et al. Exome sequencing identifies a rare HSPG2 variant associated with familial idiopathic scoliosis. G3 (Bethesda) 2014, 5, 167–174. [Google Scholar]
- Baschal, E.E.; Terhune, E.A.; Wethey, C.I.; Baschal, R.M.; Robinson, K.D.; Cuevas, M.T.; Pradhan, S.; Sutphin, B.S.; Taylor, M.R.G.; Gowan, K.; et al. Idiopathic scoliosis families highlight actin-based and microtubule-based cellular projections and extracellular matrix in disease etiology. G3 (Bethesda) 2018, 8, 2663–2672. [Google Scholar]
- Terhune, E.A.; Wethey, C.I.; Cuevas, M.T.; Monley, A.M.; Baschal, E.E.; Bland, M.R.; Baschal, R.; Trahan, G.D.; Taylor, M.R.G.; Jones, K.L.; et al. Whole exome sequencing of 23 multigeneration idiopathic scoliosis families reveals enrichments in cytoskeletal variants, suggests highly polygenic disease. Genes 2021, 12, 922. [Google Scholar] [PubMed]
- Carry, P.M.; Duke, V.R.; Brazell, C.J.; Stence, N.; Scholes, M.; Rousie, D.L.; Miller, N.H. Lateral semi-circular canal asymmetry in females with idiopathic scoliosis. PLoS ONE 2020, 15, e0232417. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernystsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar]
- Scheffer, D.I.; Shen, J.; Corey, D.P.; Chen, Z.Y. Gene expression by mouse inner ear hair cells during development. J. Neurosci. 2015, 35, 6366–6380. [Google Scholar]
- Anttila, V.; Winsvold, B.S.; Gormley, P.; Kurth, T.; Bettella, F.; McMahon, G.; Kallela, M.; Malik, R.; de Vries, B.; Terwindt, G.; et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat. Genet. 2013, 45, 912–917. [Google Scholar]
- van den Maagdenberg, A.M.J.M.; Nyholt, D.R.; Anttila, V. Novel hypotheses emerging from GWAS in migraine? J. Headache Pain 2019, 20, 5. [Google Scholar]
- Claeys, K.G.; Claes, L.R.F.; Van Goethem, J.W.M.; Sercu, S.; Merregaert, J.; Lambert, J.; Van Marck, E.A.; Parizel, P.M.; De Jonghe, P. Epilepsy and migraine in a patient with Urbach-Wiethe disease. Seizure 2007, 1, 465–468. [Google Scholar]
- Gormley, P.; Anttila, V.; Winsvold, B.S.; Palta, P.; Esko, T.; Pers, T.H.; Farh, K.H.; Cuenca-Leon, E.; Muona, M.; Furlotte, N.A.; et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 2016, 48, 856–866. [Google Scholar]
- Melo, U.S.; Macedo-Souza, L.I.; Figueiredo, T.; Muotri, A.R.; Gleeson, J.G.; Coux, G.; Armas, P.; Calcaterra, N.B.; Kitajima, J.P.; Amorim, S.; et al. Overexpression of KLC2 due to a homozygous deletion in the non-coding region causes SPOAN syndrome. Hum. Mol. Genet. 2015, 24, 6877–6885. [Google Scholar]
- Hurle, B.; Marques-Bonet, T.; Antonacci, F.; Hughes, I.; Ryan, J.F.; NISC Comparative Sequencing Program; Eichler, E.E.; Ornitz, D.M.; Green, E.D. Lineage-specific evolution of the vertebrate Otopetrin family revealed by comparative genomic analyses. BMC Evol. Biol. 2011, 11, 23. [Google Scholar]
- Wang, W.; Van De Water, T.; Lufkin, T. Inner ear and maternal reproductive defects in mice lacking the Hmx3 homeobox gene. Development 1998, 634, 621–634. [Google Scholar]
- Bahadoran, P.; Ortonne, J.P.; Ballotti, R.; de Saint-Basile, G. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am. J. Med. Genet. A 2003, 116A, 408–409. [Google Scholar]
- Joshi, G.; Pradhan, S.; Mittal, B. Vascular gene polymorphisms (EDNRA-231 G>A and APOE HhaI) and risk for migraine. DNA Cell. Biol. 2011, 30, 577–584. [Google Scholar]
- Gupta, R.; Kumar, V.; Luthra, K.; Banerjee, B.; Bhatia, M.S. Polymorphism in apolipoprotein E among migraineurs and tension-type headache subjects. J. Headache Pain 2009, 10, 115–120. [Google Scholar]
- Pillers, D.A.M.; Kempton, J.B.; Duncan, N.M.; Pang, J.; Dwinnell, S.J.; Trune, D.R. Hearing loss in the laminin-deficient dy mouse model of congenital muscular dystrophy. Mol. Genet. Metab. 2002, 76, 217–224. [Google Scholar] [PubMed]
- Hurle, B.; Ignatova, E.; Massironi, S.M.; Mashimo, T.; Rios, X.; Thalmann, I.; Thalmann, R.; Ornitz, D.M. Non-syndromic vestibular disorder with otoconial agenesis in tilted/mergulhador mice caused by mutations in otopetrin 1. Hum. Mol. Genet. 2003, 12, 777–789. [Google Scholar] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [PubMed]
- Xia, J.; Benner, M.J.; Hancock, R.E.W. NetworkAnalyst—Integrative approaches for protein–protein interaction network analysis and visual exploration. Nucleic Acids Res. 2014, 42, W167–W174. [Google Scholar]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.W.; Brinkman, F.S.L.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond—Recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [PubMed]
- Lanzi, G.; Balottin, U.; Fazzi, E.; Tagliasacchi, M.; Manfrin, M.; Mira, E. Benign paroxysmal vertigo of childhood: A long-term follow-up. Cephalalgia 1994, 14, 458–460. [Google Scholar]
- Buchan, J.G.; Gray, R.S.; Gansner, J.M.; Alvarado, D.M.; Burgert, L.; Gitlin, J.D.; Gurnett, C.A.; Goldsmith, M.I. Kinesin Family Member 6 (kif6) Is Necessary for Spine Development in Zebrafish. Dev. Dyn. 2014, 6, 1646–1657. [Google Scholar]
- Roknic, N.; Huber, A.; Hegemann, S.C.A.; Häusler, R.; Gürtler, N. Mutation analysis of Netrin 1 and HMX3 genes in patients with superior semicircular canal dehiscence syndrome. Acta Otolaryngol. 2012, 132, 1061–1065. [Google Scholar]
- Techlo, T.R.; Rasmussen, A.H.; Møller, P.L.; Bøttcher, M.; Winther, S.; Davidsson, O.B.; Olofsson, I.A.; Chalmer, M.A.; Kogelman, L.J.A.; Nyegaard, M.; et al. Familial analysis reveals rare risk variants for migraine in regulatory regions. Neurogenetics 2020, 21, 149–157. [Google Scholar]
- Tu, Y.H.; Cooper, A.J.; Teng, B.; Chang, R.B.; Artiga, D.J.; Turner, H.N.; Mulhall, E.M.; Ye, W.; Smith, A.D.; Liman, E.R. An evolutionarily conserved gene family encodes proton-selective ion channels. Science 2018, 359, 1047–1050. [Google Scholar]
- Skuladottir, A.T.; Bjornsdottir, G.; Nawaz, M.S.; Petersen, H.; Rognvaldsson, S.; Moore, K.H.S.; Olafsson, O.A.; Sigurdardottir, G.R.; Saevarsdottir, S.; Ivarsdottir, E.V.; et al. A genome-wide meta-analysis uncovers six sequence variants conferring risk of vertigo. Commun. Biol. 2021, 4, 1148. [Google Scholar]
- Simoneau, M.; Lamothe, V.; Hutin, E.; Mercier, P.; Teasdale, N.; Blouin, J. Evidence for cognitive vestibular integration impairment in idiopathic scoliosis patients. BMC Neurosci. 2009, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chau, W.W.; Hui-Chan, C.W.Y.; Cheung, C.S.K.; Tsang, W.W.N.; Cheng, J.C.Y. Balance control in adolescents with idiopathic scoliosis and disturbed somatosensory function. Spine J. 2006, 31, 437–440. [Google Scholar]
- Miller, N.D.; Nance, M.A.; Wohler, E.S.; Hoover-Fong, J.E.; Lisi, E.; Thomas, G.H.; Pevsner, J. Molecular (SNP) Analyses of Overlapping Hemizygous Deletions of 10q25. 3 to 10qter in Four Patients: Evidence for HMX2 and HMX3 as Candidate Genes in Hearing and Vestibular Function. Am. J. Med. Genet. A 2009, 149A, 669–680. [Google Scholar]
- Oliveira, J.; Gruber, A.; Cardoso, M.; Taipa, R.; Fineza, I.; Gonçalves, A.; Laner, A.; Winder, T.L.; Schroeder, J.; Rath, J.; et al. LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum. Mutat. 2018, 39, 1314–1337. [Google Scholar]
- Geranmayeh, F.; Clement, E.; Feng, L.H.; Sewry, C.; Pagan, J.; Mein, R.; Abbs, S.; Brueton, L.; Childs, A.M.; Jungbluth, H.; et al. Neuromuscular Disorders Genotype—Phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul. Disord. 2010, 20, 241–250. [Google Scholar]
- Schmahmann, J.D.; Weilburg, J.B.; Sherman, J.C. The neuropsychiatry of the cerebellum—Insights from the clinic. Cerebellum 2007, 6, 254–267. [Google Scholar]
- Kayan, A.; Hood, J.D. Neuro-otological Manifestations of Migraine. Brain 1984, 107, 1123–1142. [Google Scholar] [CrossRef]
- Neuhauser, H.; Leopold, M.; von Brevern, M.; Arnold, G.; Lempert, T. The interrelations of migraine, vertigo, and migrainous vertigo. Neurology 2001, 56, 436–441. [Google Scholar] [CrossRef]
- Neuhauser, H.K.; von Brevern, M.; Radtke, A.; Lezius, F.; Feldmann, M.; Ziese, T.; Lempert, T. Epidemiology of vestibular vertigo: A neurotologic survey of the general population. Neurology 2005, 65, 898–904. [Google Scholar]
- Cavestro, C.; Mandrino, S. Thrombophilic disorders in migraine. Front. Neurol. 2014, 5, 120. [Google Scholar] [CrossRef]
- Tietjen, G.E.; Collins, S.A. Hypercoagulability and Migraine. Headache 2018, 58, 173–183. [Google Scholar] [PubMed]
- Fattori, B.; Nacci, A.; Casani, A.; Cristofani, R.; Sagripanti, A. Hemostatic alterations in patients with acute, unilateral vestibular paresis. Otolaryngol. Head Neck Surg. 2001, 124, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Skarp, S.; Kanervo, L.; Kotimäki, J.; Sorri, M.; Männikkö, M.; Hietikko, E. Whole-exome sequencing suggests multiallelic inheritance for childhood-onset Ménière’s disease. Ann. Hum. Genet. 2019, 83, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sierra, C.; Gallego-Martinez, A.; Requena, T.; Frejo, L.; Batuecas-Caletrío, A.; Lopez-Escamez, J.A. Variable expressivity and genetic heterogeneity involving DPT and SEMA3D genes in autosomal dominant familial Meniere’s disease. Eur. J. Hum. Genet. 2017, 25, 200–207. [Google Scholar]
- Roman-Naranjo, P.; Gallego-Martinez, A.; Soto-Varela, A.; Aran, I.; Moleon, M.D.C.; Espinosa-Sanchez, J.M.; Amor-Dorado, J.C.; Batuecas-Caletrio, A.; Perez-Vazquez, P.; Lopez-Escamez, J.A. Burden of rare variants in the OTOG gene in Familial Meniere’s Disease. Ear. Hear. 2020, 41, 1598–1605. [Google Scholar]
- Roman-Naranjo, P.; Moleon, M.D.C.; Aran, I.; Escalera-Balsera, A.; Soto-Varela, A.; Bächinger, D.; Gomez-Fiñana, M.; Eckhard, A.H.; Lopez-Escamez, J.A. Rare coding variants involving MYO7A and other genes encoding stereocilia link proteins in familial Meniere Disease. Hear. Res. 2021, 409, 108329. [Google Scholar]
- Kentala, E.; Rauch, S.D. A practical assessment algorithm for diagnosis of dizziness. Otolaryngol. Head Neck Surg. 2003, 128, 54–59. [Google Scholar]


| PANTHER (Biological Processes) | Gene Ontology (Biological Processes) | REACTOME | |||
|---|---|---|---|---|---|
| Pathway | FDR-adj-p | Pathway | FDR-adj-p | Pathway | FDR-adj-p |
| Receptor-mediated endocytosis | 2.60 × 10–7 | Vesicle-mediated transport | 1.19 × 10–10 | Activation of BH3-only proteins | 8.86 × 10–13 |
| Regulation of binding | 8.70 × 10–6 | Coagulation | 1.71× 10–10 | Intrinsic pathway for apoptosis | 1.33 × 10–10 |
| Protein targeting | 2.10 × 10–5 | Transcription initiation from RNA polymerase II promoter | 4.07 × 10–10 | Translocation of GLUT4 to the plasma membrane | 6.39 × 10–10 |
| Viral process | 2.20 × 10–5 | Blood coagulation | 6.91 × 10–10 | Activation of BAD and translocation to mitochondria | 3.11 × 10–9 |
| Negative regulation of apoptotic process | 6.30 × 10–5 | Hemostasis | 8.77 × 10–10 | Membrane trafficking | 1.11 × 10–8 |
| Chemical synaptic transmission | 1.30 × 10–4 | Regulation of body fluid levels | 2.59 × 10–9 | Hemostasis | 4.02 × 10–8 |
| Cholesterol metabolic process | 1.10 × 10–3 | Wound healing | 5.08 × 10–9 | Platelet degranulation | 9.93 × 10–7 |
| Endocytosis | 2.90 × 10–3 | DNA-dependent transcription, initiation | 5.30 × 10–9 | Platelet activation, signaling, and aggregation | 1.29 × 10–6 |
| Heart development | 4.30 × 10–3 | Exocytosis | 1.64 × 10–8 | Insulin processing | 1.32 × 10–6 |
| Vitamin metabolic process | 4.50 × 10–3 | Cell morphogenesis involved in differentiation | 1.71 × 10–8 | Response to elevated platelet cytosolic Ca2+ | 1.65 × 10–6 |
| Protein localization | 1.26 × 10–6 | ||||
| ID | Gene | Variant | Highest MAF | Damaging Prediction | Scaled CADD |
|---|---|---|---|---|---|
| S19 | ECM1 | c.844C>T (p.(Arg282Trp) | (AU)Lat: 1.3 × 10–4 | PR, SI | 24.6 |
| 60 | OTOP1 | c.380A>T (p.(His127Leu)) | (AU)Eur: 7.3 × 10–5 | PP, MT | 22.8 |
| 9 | OTOP2 | c.760G>A (p.(Ala254Thr)) | (AU)Afr: 2.2 × 10–4 | PP, MT | 23.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumalde, A.A.M.; Scholes, M.A.; Kalmanson, O.A.; Terhune, E.A.; Frejo, L.; Wethey, C.I.; Roman-Naranjo, P.; Carry, P.M.; Gubbels, S.P.; Lopez-Escamez, J.A.; et al. Rare Coding Variants in Patients with Non-Syndromic Vestibular Dysfunction. Genes 2023, 14, 831. https://doi.org/10.3390/genes14040831
Sumalde AAM, Scholes MA, Kalmanson OA, Terhune EA, Frejo L, Wethey CI, Roman-Naranjo P, Carry PM, Gubbels SP, Lopez-Escamez JA, et al. Rare Coding Variants in Patients with Non-Syndromic Vestibular Dysfunction. Genes. 2023; 14(4):831. https://doi.org/10.3390/genes14040831
Chicago/Turabian StyleSumalde, Angelo Augusto M., Melissa A. Scholes, Olivia A. Kalmanson, Elizabeth A. Terhune, Lidia Frejo, Cambria I. Wethey, Pablo Roman-Naranjo, Patrick M. Carry, Samuel P. Gubbels, Jose A. Lopez-Escamez, and et al. 2023. "Rare Coding Variants in Patients with Non-Syndromic Vestibular Dysfunction" Genes 14, no. 4: 831. https://doi.org/10.3390/genes14040831
APA StyleSumalde, A. A. M., Scholes, M. A., Kalmanson, O. A., Terhune, E. A., Frejo, L., Wethey, C. I., Roman-Naranjo, P., Carry, P. M., Gubbels, S. P., Lopez-Escamez, J. A., Hadley-Miller, N., & Santos-Cortez, R. L. P. (2023). Rare Coding Variants in Patients with Non-Syndromic Vestibular Dysfunction. Genes, 14(4), 831. https://doi.org/10.3390/genes14040831