
Insight into the Natural History of Pathogenic Variant c.919-2A>G in the SLC26A4 Gene Involved in Hearing Loss: The Evidence for Its Common Origin in Southern Siberia (Russia)

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Ethics Statement
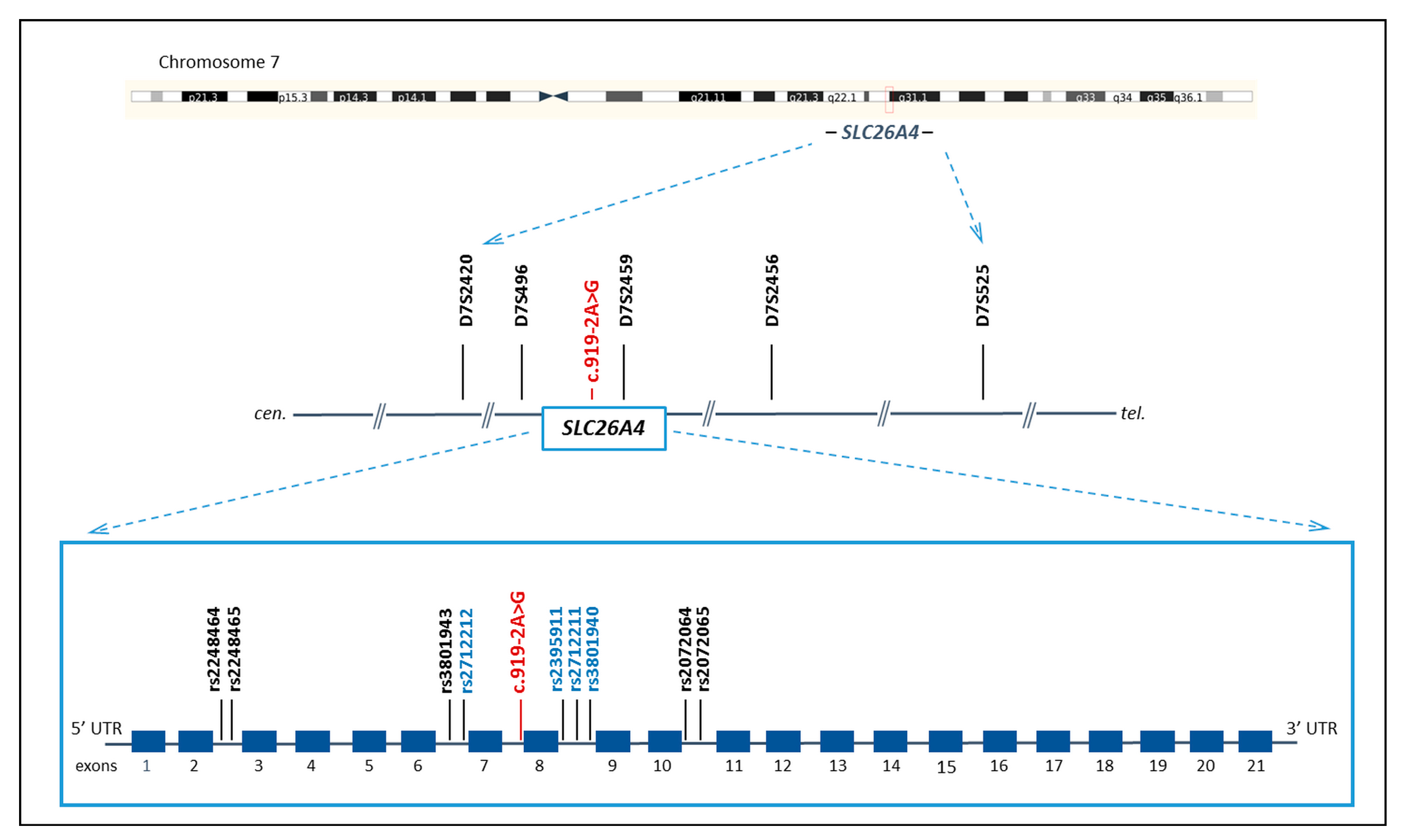
2.3. STRs and SNPs Genotyping
2.4. Reconstruction of STR and SNP Haplotypes
2.5. Estimation of c.919-2A>G Age
2.6. Statistical Analysis
3. Results
3.1. STR and SNP Haplotypes
3.2. Estimation of c.919-2A>G Age
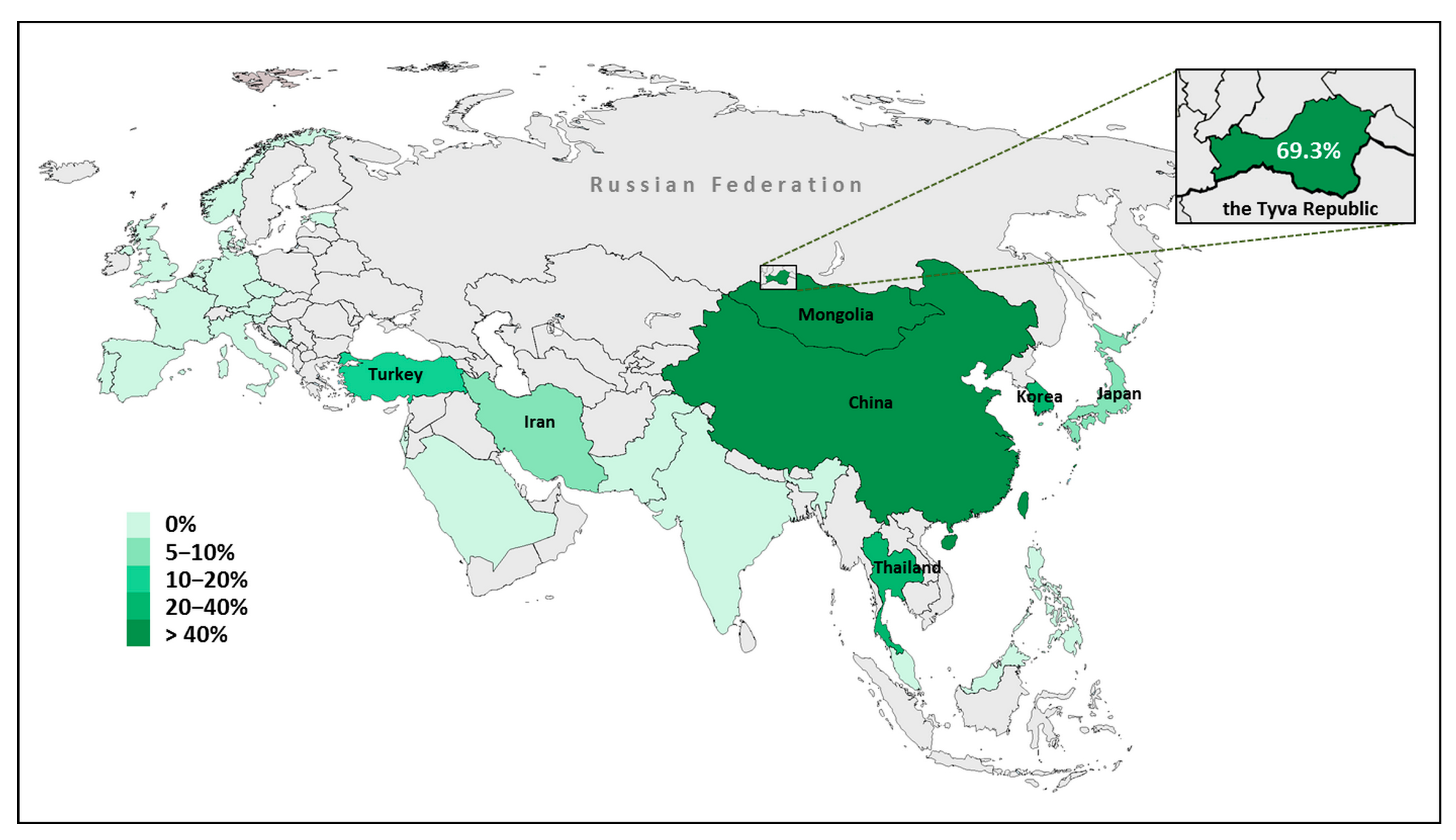
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Everett, L.A.; Glaser, B.; Beck, J.C.; Idol, J.R.; Buchs, A.; Heyman, M.; Adawi, F.; Hazani, E.; Nassir, E.; Baxevanis, A.D.; et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat. Genet. 1997, 17, 411–422. [Google Scholar] [CrossRef]
- Everett, L.A.; Morsli, H.; Wu, D.K.; Green, E.D. Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc. Natl. Acad. Sci. USA 1999, 96, 9727–9732. [Google Scholar] [CrossRef]
- Honda, K.; Griffith, A.J. Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss. Hum. Genet. 2022, 141, 455–464. [Google Scholar] [CrossRef]
- Albert, S.; Blons, H.; Jonard, L.; Feldmann, D.; Chauvin, P.; Loundon, N.; Sergent-Allaoui, A.; Houang, M.; Joannard, A.; Schmerber, S.; et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur. J. Hum. Genet. 2006, 14, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Guo, Y.; Wang, C.; Wang, Y.; Liu, X. A systematic review and meta-analysis of common mutations of SLC26A4 gene in Asian populations. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Nishio, S.Y.; Usami, S.; Deafness Gene Study Consortium. Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: A large cohort study. J. Hum. Genet. 2014, 59, 262–268. [Google Scholar] [CrossRef]
- Lu, Y.J.; Yao, J.; Wei, Q.J.; Xing, G.Q.; Cao, X. Diagnostic Value of SLC26A4 Mutation Status in Hereditary Hearing Loss With EVA: A PRISMA-Compliant Meta-Analysis. Medicine 2015, 94, e2248. [Google Scholar] [CrossRef]
- Tsukada, K.; Nishio, S.Y.; Hattori, M.; Usami, S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. S1), 61S–76S. [Google Scholar] [CrossRef]
- Koohiyan, M. A systematic review of SLC26A4 mutations causing hearing loss in the Iranian population. Int. J. Pediatr. Otorhinolaryngol. 2019, 125, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Naz, S. Molecular genetic landscape of hereditary hearing loss in Pakistan. Hum. Genet. 2022, 141, 633–648. [Google Scholar] [CrossRef]
- Park, H.J.; Shaukat, S.; Liu, X.Z.; Hahn, S.H.; Naz, S.; Ghosh, M.; Kim, H.N.; Moon, S.K.; Abe, S.; Tukamoto, K.; et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: Global implications for the epidemiology of deafness. J. Med. Genet. 2003, 40, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Yeh, T.H.; Chen, P.J.; Hsu, C.J. Prevalent SLC26A4 mutations in patients with enlarged vestibular aqueduct and/or Mondini dysplasia: A unique spectrum of mutations in Taiwan, including a frequent founder mutation. Laryngoscope 2005, 115, 1060–1064. [Google Scholar] [CrossRef]
- Borck, G.; Roth, C.; Martiné, U.; Wildhardt, G.; Pohlenz, J. Mutations in the PDS gene in German families with Pendred’s syndrome: V138F is a founder mutation. J. Clin. Endocrinol. Metab. 2003, 88, 2916–2921. [Google Scholar] [CrossRef]
- Pera, A.; Dossena, S.; Rodighiero, S.; Gandía, M.; Bottà, G.; Meyer, G.; Moreno, F.; Nofziger, C.; Hernández-Chico, C.; Paulmichl, M. Functional assessment of allelic variants in the SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA. Proc. Natl. Acad. Sci. USA 2008, 105, 18608–18613. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, M.; Honarpour, A.; Mozafari, R.; Davarnia, B.; Najmabadi, H.; Kahrizi, K. Identification of a founder mutation for Pendred syndrome in families from northwest Iran. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1828–1832. [Google Scholar] [CrossRef]
- Anwar, S.; Riazuddin, S.; Ahmed, Z.M.; Tasneem, S.; Ateeq-ul-Jaleel; Khan, S.Y.; Griffith, A.J.; Friedman, T.B.; Riazuddin, S. SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred’s syndrome in Pakistanis. J. Hum. Genet. 2009, 54, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Coucke, P.J.; van Hauwe, P.; Everett, L.A.; Demirhan, O.; Kabakkaya, Y.; Dietrich, N.L.; Smith, R.J.; Coyle, E.; Reardon, W.; Trembath, R.; et al. Identification of two different mutations in the PDS gene in an inbred family with Pendred syndrome. J. Med. Genet. 1999, 36, 475–477. [Google Scholar] [PubMed]
- Yang, J.J.; Tsai, C.C.; Hsu, H.M.; Shiao, J.Y.; Su, C.C.; Li, S.Y. Hearing loss associated with enlarged vestibular aqueduct and Mondini dysplasia is caused by splice-site mutation in the PDS gene. Hear Res. 2005, 199, 22–30. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Suzuki, H.; Harada, D.; Namba, A.; Abe, S.; Usami, S. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: A unique spectrum of mutations in Japanese. Eur. J. Hum. Genet. 2003, 11, 916–922. [Google Scholar] [CrossRef]
- Wang, Q.J.; Zhao, Y.L.; Rao, S.Q.; Guo, Y.F.; Yuan, H.; Zong, L.; Guan, J.; Xu, B.C.; Wang, D.Y.; Han, M.K.; et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin. Genet. 2007, 72, 245–254. [Google Scholar] [CrossRef]
- Dai, P.; Li, Q.; Huang, D.; Yuan, Y.; Kang, D.; Miller, D.T.; Shao, H.; Zhu, Q.; He, J.; Yu, F.; et al. SLC26A4 c.919-2A>G varies among Chinese ethnic groups as a cause of hearing loss. Genet. Med. 2008, 10, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.L.; Bai-Cheng, X.; Chen, X.J.; Pan-Pan, B.; Jian-Li, M.; Xiao-Wen, L.; Zhang, Z.W.; Wan, D.; Zhu, Y.M.; Guo, Y.F. Common molecular etiology of patients with nonsyndromic hearing loss in Tibetan, Tu nationality, and Mongolian patients in the northwest of China. Acta Otolaryngol. 2013, 133, 930–934. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, J.; Shin, J.W.; Song, M.H.; Kim, S.H.; Lee, J.H.; Lee, K.A.; Shin, S.; Kim, U.K.; Bok, J.; et al. Correlation between genotype and phenotype in patients with bi-allelic SLC26A4 mutations. Clin. Genet. 2014, 86, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ao, L.; Ding, H.; Zhang, D. Genetic frequencies related to severe or profound sensorineural hearing loss in Inner Mongolia Autonomous Region. Genet. Mol. Biol. 2016, 39, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Erdenechuluun, J.; Lin, Y.-H.; Ganbat, K.; Bataakhuu, D.; Makhbal, Z.; Tsai, C.-Y.; Lin, Y.-H.; Chan, Y.-H.; Hsu, C.-J.; Hsu, W.-C.; et al. Unique spectra of deafness-associated mutations in Mongolians provide insights into the genetic relationships among Eurasian populations. PLoS ONE 2018, 13, e0209797. [Google Scholar] [CrossRef]
- Wu, C.C.; Tsai, C.Y.; Lin, Y.H.; Chen, P.Y.; Lin, P.H.; Cheng, Y.F.; Wu, C.M.; Lin, Y.H.; Lee, C.Y.; Erdenechuluun, J.; et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes 2019, 10, 772. [Google Scholar] [CrossRef]
- Danilchenko, V.Y.; Zytsar, M.V.; Maslova, E.A.; Bady-Khoo, M.S.; Barashkov, N.A.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L. Different Rates of the SLC26A4-Related Hearing Loss in Two Indigenous Peoples of Southern Siberia (Russia). Diagnostics 2021, 11, 2378. [Google Scholar] [CrossRef]
- Baldwin, C.T.; Weiss, S.; Farrer, L.A.; De Stefano, A.L.; Adair, R.; Franklyn, B.; Kidd, K.K.; Korostishevsky, M.; Bonné-Tamir, B. Linkage of congenital, recessive deafness (DFNB4) to chromosome 7q31 and evidence for genetic heterogeneity in the Middle Eastern Druze population. Hum. Mol. Genet. 1995, 4, 1637–1642. [Google Scholar] [CrossRef]
- Coucke, P.; Van Camp, G.; Demirhan, O.; Kabakkaya, Y.; Balemans, W.; Van Hauwe, P.; Van Agtmael, T.; Smith, R.J.; Parving, A.; Bolder, C.H.; et al. The gene for Pendred syndrome is located between D7S501 and D7S692 in a 1.7-cM region on chromosome 7q. Genomics 1997, 40, 48–54. [Google Scholar] [CrossRef]
- Gausden, E.; Coyle, B.; Armour, J.A.; Coffey, R.; Grossman, A.; Fraser, G.R.; Winter, R.M.; Pembrey, M.E.; Kendall-Taylor, P.; Stephens, D.; et al. Pendred syndrome: Evidence for genetic homogeneity and further refinement of linkage. J. Med. Genet. 1997, 34, 126–129. [Google Scholar] [CrossRef]
- López-Bigas, N.; Rabionet, R.; de Cid, R.; Govea, N.; Gasparini, P.; Zelante, L.; Arbonés, M.L.; Estivill, X. Splice-site mutation in the PDS gene may result in intrafamilial variability for deafness in Pendred syndrome. Hum. Mutat. 1999, 14, 520–526. [Google Scholar] [CrossRef]
- Gonzalez Trevino, O.; Karamanoglu Arseven, O.; Ceballos, C.J.; Vives, V.I.; Ramirez, R.C.; Gomez, V.V.; Medeiros-Neto, G.; Kopp, P. Clinical and molecular analysis of three Mexican families with Pendred’s syndrome. Eur. J. Endocrinol. 2001, 144, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Yazdanpanahi, N.; Tabatabaiefar, M.A.; Bagheri, N.; Azadegan Dehkordi, F.; Farrokhi, E.; Hashemzadeh Chaleshtori, M. The role and spectrum of SLC26A4 mutations in Iranian patients with autosomal recessive hereditary deafness. Int. J. Audiol. 2015, 54, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Bengtsson, B.O.; Thomson, G. Measuring the strength of associations between HLA antigens and diseases. Tissue Antigens 1981, 18, 356–363. [Google Scholar] [CrossRef]
- Rannala, B.; Bertorelle, G. Using linked markers to infer the age of a mutation. Hum. Mutat. 2001, 18, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Risch, N.; de Leon, D.; Ozelius, L.; Kramer, P.; Almasy, L.; Singer, B.; Fahn, S.; Breakefield, X.; Bressman, S. Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat. Genet. 1995, 9, 152–159. [Google Scholar] [CrossRef]
- Nonose, R.W.; Lezirovitz, K.; de Mello Auricchio, M.T.B.; Batissoco, A.C.; Yamamoto, G.L.; Mingroni-Netto, R.C. Mutation analysis of SLC26A4 (Pendrin) gene in a Brazilian sample of hearing-impaired subjects. BMC Med. Genet. 2018, 19, 73. [Google Scholar] [CrossRef]
- Pryor, S.P.; Madeo, A.C.; Reynolds, J.C.; Sarlis, N.J.; Arnos, K.S.; Nance, W.E.; Yang, Y.; Zalewski, C.K.; Brewer, C.C.; Butman, J.A.; et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): Evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J. Med. Genet. 2005, 42, 159–165. [Google Scholar] [CrossRef]
- Dai, P.; Stewart, A.K.; Chebib, F.; Hsu, A.; Rozenfeld, J.; Huang, D.; Kang, D.; Lip, V.; Fang, H.; Shao, H.; et al. Distinct and novel SLC26A4/Pendrin mutations in Chinese and U.S. patients with nonsyndromic hearing loss. Physiol. Genom. 2009, 38, 281–290. [Google Scholar] [CrossRef]
- Dahl, H.H.; Ching, T.Y.; Hutchison, W.; Hou, S.; Seeto, M.; Sjahalam-King, J. Etiology and audiological outcomes at 3 years for 364 children in Australia. PLoS ONE 2013, 8, e59624. [Google Scholar] [CrossRef] [PubMed]
- de Moraes, V.C.; dos Santos, N.Z.; Ramos, P.Z.; Svidnicki, M.C.; Castilho, A.M.; Sartorato, E.L. Molecular analysis of SLC26A4 gene in patients with nonsyndromic hearing loss and EVA: Identification of two novel mutations in Brazilian patients. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.D.C.E.S.; Grangeiro, C.H.P.; Picanço-Albuquerque, C.G.; Dos Anjos, T.O.; De Molfetta, G.A.; Silva, W.A., Jr.; Ferraz, V.E.F. Contribution of SLC26A4 to the molecular diagnosis of nonsyndromic prelingual sensorineural hearing loss in a Brazilian cohort. BMC Res. Notes 2018, 11, 546. [Google Scholar] [CrossRef] [PubMed]
- Talbi, S.; Bonnet, C.; Riahi, Z.; Boudjenah, F.; Dahmani, M.; Hardelin, J.P.; Wong Jun Tai, F.; Louha, M.; Ammar-Khodja, F.; Petit, C. Genetic heterogeneity of congenital hearing impairment in Algerians from the Ghardaïa province. Int. J. Pediatr. Otorhinolaryngol. 2018, 112, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Torre-González, C.; Villanueva-García, D.; García-Delgado, C.; Castillo-Castillo, S.; Huante-Guido, M.; Chichitz-Madrigal, J.; Juárez-Torres, M.E.; Sánchez-Sandoval, A.L.; Barrón-Palma, E.V.; Morán-Barroso, V.F. Congenital hearing loss: A literature review of the genetic etiology in a Mexican population. Bol. Med. Hosp. Infant. Mex. 2022, 79, 206–214. [Google Scholar] [CrossRef]
- Wonkam, A.; Adadey, S.M.; Schrauwen, I.; Aboagye, E.T.; Wonkam-Tingang, E.; Esoh, K.; Popel, K.; Manyisa, N.; Jonas, M.; deKock, C.; et al. Exome sequencing of families from Ghana reveals known and candidate hearing impairment genes. Commun. Biol. 2022, 5, 369. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Guo, W.; Tang, J.; Zhang, G.; Wang, G.; Han, M.; Zhang, X.; Yang, S.; He, D.Z.; Dai, P. Molecular epidemiology and functional assessment of novel allelic variants of SLC26A4 in non-syndromic hearing loss patients with enlarged vestibular aqueduct in China. PLoS ONE 2012, 7, e49984. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Yuan, Y.; Huang, D.; Zhu, X.; Yu, F.; Kang, D.; Yuan, H.; Wu, B.; Han, D.; Wong, L.J. Molecular etiology of hearing impairment in Inner Mongolia: Mutations in SLC26A4 gene and relevant phenotype analysis. J. Transl. Med. 2008, 6, 74. [Google Scholar] [CrossRef]
- Chai, Y.; Huang, Z.; Tao, Z.; Li, X.; Li, L.; Li, Y.; Wu, H.; Yang, T. Molecular etiology of hearing impairment associated with nonsyndromic enlarged vestibular aqueduct in East China. Am. J. Med. Genet. A 2013, 161A, 2226–2233. [Google Scholar] [CrossRef]
- Xin, F.; Yuan, Y.; Deng, X.; Han, M.; Wang, G.; Zhao, J.; Gao, X.; Liu, J.; Yu, F.; Han, D.; et al. Genetic mutations in nonsyndromic deafness patients of Chinese minority and Han ethnicities in Yunnan, China. J. Transl. Med. 2013, 11, 312. [Google Scholar] [CrossRef]
- Chen, K.; Zong, L.; Liu, M.; Wang, X.; Zhou, W.; Zhan, Y.; Cao, H.; Dong, C.; Tang, H.; Jiang, H. Developing regional genetic counseling for southern Chinese with nonsyndromic hearing impairment: A unique mutational spectrum. J. Transl. Med. 2014, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Wang, Q.; Zhu, Y.; Wang, Y.; Guo, Y. Associations between GJB2, mitochondrial 12S rRNA, SLC26A4 mutations, and hearing loss among three ethnicities. Biomed. Res. Int. 2014, 2014, 746838. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.H.; Zhu, Y.M.; Wang, Y.L.; Guo, Y.F. Common molecular etiology of nonsyndromic hearing loss in 484 patients of 3 ethnicities in northwest China. Acta Otolaryngol. 2015, 135, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Han, M.; Wang, G.; Huang, S.; Zeng, J.; Yuan, Y.; Dai, P. Genetic mutations in non-syndromic deafness patients in Hainan Province have a different mutational spectrum compared to patients from Mainland China. Int. J. Pediatr. Otorhinolaryngol. 2018, 108, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, Y.; Wu, J.; Chen, S.; Tang, S.; Jiang, Y.; Dai, P. Study on the relationship between the pathogenic mutations of SLC26A4 and CT phenotypes of inner ear in patient with sensorineural hearing loss. Biosci. Rep. 2019, 39, BSR20182241. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, S.J.; Jin, H.S.; Lee, J.O.; Go, S.H.; Jang, H.S.; Moon, S.K.; Lee, S.C.; Chun, Y.M.; Lee, H.K.; et al. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin. Genet. 2005, 67, 160–165. [Google Scholar] [CrossRef]
- Rah, Y.C.; Kim, A.R.; Koo, J.W.; Lee, J.H.; Oh, S.H.; Choi, B.Y. Audiologic presentation of enlargement of the vestibular aqueduct according to the SLC26A4 genotypes. Laryngoscope 2015, 125, E216–E222. [Google Scholar] [CrossRef]
- Snabboon, T.; Plengpanich, W.; Saengpanich, S.; Sirisalipoch, S.; Keelawat, S.; Sunthornyothin, S.; Khovidhunkit, W.; Suwanwalaikorn, S.; Sridama, V.; Shotelersuk, V. Two common and three novel PDS mutations in Thai patients with Pendred syndrome. J. Endocrinol. Investig. 2007, 30, 907–913. [Google Scholar] [CrossRef]
- Cengiz, F.B.; Yilmazer, R.; Olgun, L.; Sennaroglu, L.; Kirazli, T.; Alper, H.; Olgun, Y.; Incesulu, A.; Atik, T.; Huesca-Hernandez, F.; et al. Novel pathogenic variants underlie SLC26A4-related hearing loss in a multiethnic cohort. Int. J. Pediatr. Otorhinolaryngol. 2017, 101, 167–171. [Google Scholar] [CrossRef]
- Reiisi, S.; Sanati, M.H.; Tabatabaiefar, M.A.; Ahmadian, S.; Reiisi, S.; Parchami, S.; Porjafari, H.; Shahi, H.; Shavarzi, A.; Hashemzade Chaleshtori, M. The Study of SLC26A4 Gene Causing Autosomal Recessive Hearing Loss by Linkage Analysis in a Cohort of Iranian Populations. Int. J. Mol. Cell. Med. 2014, 3, 176–182. [Google Scholar]
- van Hauwe, P.; Everett, L.A.; Coucke, P.; Scott, D.A.; Kraft, M.L.; Ris-Stalpers, C.; Bolder, C.; Otten, B.; de Vijlder, J.J.; Dietrich, N.L.; et al. Two frequent missense mutations in Pendred syndrome. Hum. Mol. Genet. 1998, 7, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Rendtorff, N.D.; Schrijver, I.; Lodahl, M.; Rodriguez-Paris, J.; Johnsen, T.; Hansén, E.C.; Nickelsen, L.A.; Tümer, Z.; Fagerheim, T.; Wetke, R.; et al. SLC26A4 mutation frequency and spectrum in 109 Danish Pendred syndrome/DFNB4 probands and a report of nine novel mutations. Clin. Genet. 2013, 84, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Azadegan-Dehkordi, F.; Ahmadi, R.; Bahrami, T.; Yazdanpanahi, N.; Farrokhi, E.; Tabatabaiefar, M.A.; Hashemzadeh-Chaleshtori, M. A novel variant of SLC26A4 and first report of the c.716T>A variant in Iranian pedigrees with non-syndromic sensorineural hearing loss. Am. J. Otolaryngol. 2018, 39, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Mojtabavi Naeini, M.; Mesrian Tanha, H.; Hashemzadeh Chaleshtori, M.; Vallian, S. Genotyping data and novel haplotype diversity of STR markers in the SLC26A4 gene region in five ethnic groups of the Iranian population. Genet. Test. Mol. Biomarkers 2014, 18, 820–825. [Google Scholar] [CrossRef]
- Slatkin, M.; Rannala, B. Estimating allele age. Annu. Rev. Genom. Hum. Genet. 2000, 1, 225–249. [Google Scholar] [CrossRef]
- Labuda, D.; Zietkiewicz, E.; Labuda, M. The genetic clock and the age of the founder effect in growing populations: A lesson from French Canadians and Ashkenazim. Am. J. Hum. Genet. 1997, 61, 768–771. [Google Scholar] [CrossRef]
- Colombo, R. Age estimate of the N370S mutation causing Gaucher disease in Ashkenazi Jews and European populations: A reappraisal of haplotype data. Am. J. Hum. Genet. 2000, 66, 692–697. [Google Scholar] [CrossRef]
- Mongush, M.V. Tuvans of Mongolia and China. Int. J. Cent. Asian Stud. 1996, 1, 225–243. [Google Scholar]
- Chen, Z.; Zhang, Y.; Fan, A.; Zhang, Y.; Wu, Y.; Zhao, Q.; Zhou, Y.; Zhou, C.; Bawudong, M.; Mao, X.; et al. Brief communication: Y-chromosome haplogroup analysis indicates that Chinese Tuvans share distinctive affinity with Siberian Tuvans. Am. J. Phys. Anthropol. 2011, 144, 492–497. [Google Scholar] [CrossRef]
- Vainshtein, S.I.; Mannay-Ool, M.H. History of Tyva, 2nd ed.; Science: Novosibirsk, Russia, 2001. (In Russian) [Google Scholar]
- Mannai-ool, M.K. Tuvan People. The Origin and Formation of the Ethnos; Nauka Publ.: Novosibirsk, Russia, 2004; pp. 99–166. (In Russian) [Google Scholar]



{kind=link}
{kind=link}
| STR Haplotypes D7S2420-D7S496-/c.919-2A>G/-D7S2459-D7S2456-D7S525 (~2.8 Mb) | Frequency of Haplotypes | X2 | p | |
|---|---|---|---|---|
| Mutant Chromosomes | Normal Chromosomes | |||
| 278-120-147-244-227 | 0.9130 | 0.0 | 150 | <10−35 |
| 278-120-147-244-229 | 0.0435 | 0.0 | 2.4 | 0.0704 |
| 278-120-147-244-221 | 0.0217 | 0.0 | 0.28 | 0.2674 |
| 278-120-147-244-225 | 0.0217 | 0.0 | 0.28 | 0.2674 |
| Other haplotypes | 0.0 | 1.0 | - | - |
| SNP Haplotypes rs2248464-rs2248465-rs3801943-rs2712212*-/c.919-2A>G/-rs2395911*-rs2712211*-rs3801940*-rs2072064-rs2072065 (31.039 kb) | Frequency of Haplotypes | X2 | p | |
| Mutant Chromosomes | Normal Chromosomes | |||
| A-C-T-A-G-G-C-A-C | 1.0 | 0.0280 | 150 | <10−36 |
| Other haplotypes | 0.0 | 0.9720 | - | - |
| Genetic Markers Used for Calculations | d | The Single-Marker Method | The DMLE + Calculation | ||
|---|---|---|---|---|---|
| g | Age | g (95% CI) | Age (95% CI) | ||
| STR markers * | 0.05 | 22 | 550 years | 103–198 | 2575–4950 years |
| 0.1 | 21 | 525 years | 63–107 | 1575–2675 years | |
| 0.2 | 17 | 425 years | 35–59 | 875–1475 years | |
| SNP markers | 0.05 | - | - | 91–191 | 2275–4775 years |
| 0.1 | 53–103 | 1325–2575 years | |||
| 0.2 | 29–54 | 725–1350 years | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danilchenko, V.Y.; Zytsar, M.V.; Maslova, E.A.; Orishchenko, K.E.; Posukh, O.L. Insight into the Natural History of Pathogenic Variant c.919-2A>G in the SLC26A4 Gene Involved in Hearing Loss: The Evidence for Its Common Origin in Southern Siberia (Russia). Genes 2023, 14, 928. https://doi.org/10.3390/genes14040928
Danilchenko VY, Zytsar MV, Maslova EA, Orishchenko KE, Posukh OL. Insight into the Natural History of Pathogenic Variant c.919-2A>G in the SLC26A4 Gene Involved in Hearing Loss: The Evidence for Its Common Origin in Southern Siberia (Russia). Genes. 2023; 14(4):928. https://doi.org/10.3390/genes14040928
Chicago/Turabian StyleDanilchenko, Valeriia Yu., Marina V. Zytsar, Ekaterina A. Maslova, Konstantin E. Orishchenko, and Olga L. Posukh. 2023. "Insight into the Natural History of Pathogenic Variant c.919-2A>G in the SLC26A4 Gene Involved in Hearing Loss: The Evidence for Its Common Origin in Southern Siberia (Russia)" Genes 14, no. 4: 928. https://doi.org/10.3390/genes14040928
APA StyleDanilchenko, V. Y., Zytsar, M. V., Maslova, E. A., Orishchenko, K. E., & Posukh, O. L. (2023). Insight into the Natural History of Pathogenic Variant c.919-2A>G in the SLC26A4 Gene Involved in Hearing Loss: The Evidence for Its Common Origin in Southern Siberia (Russia). Genes, 14(4), 928. https://doi.org/10.3390/genes14040928