
Historical Mitogenomic Diversity and Population Structuring of Southern Hemisphere Fin Whales
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
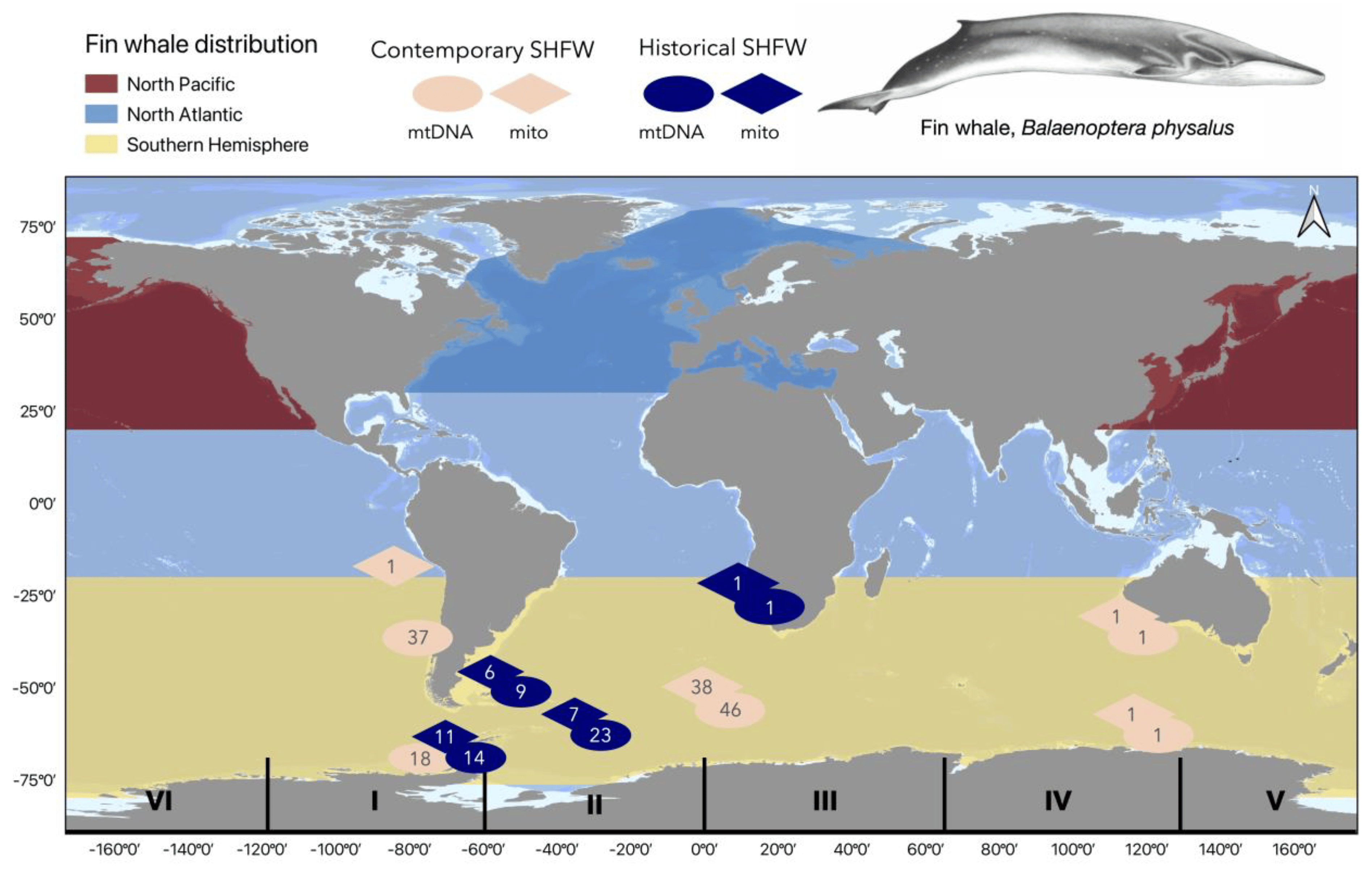
2.1. Sample Collection and DNA Extraction
2.2. mtDNA Control Region Sequencing
2.3. Next-Generation Sequencing
2.4. Bioinformatics and Mitogenome Assembly
2.5. SHFW Diversity and Population Structure
2.6. Comparison with Northern Hemisphere Fin Whales
2.7. Phylogenetic Analysis
3. Results
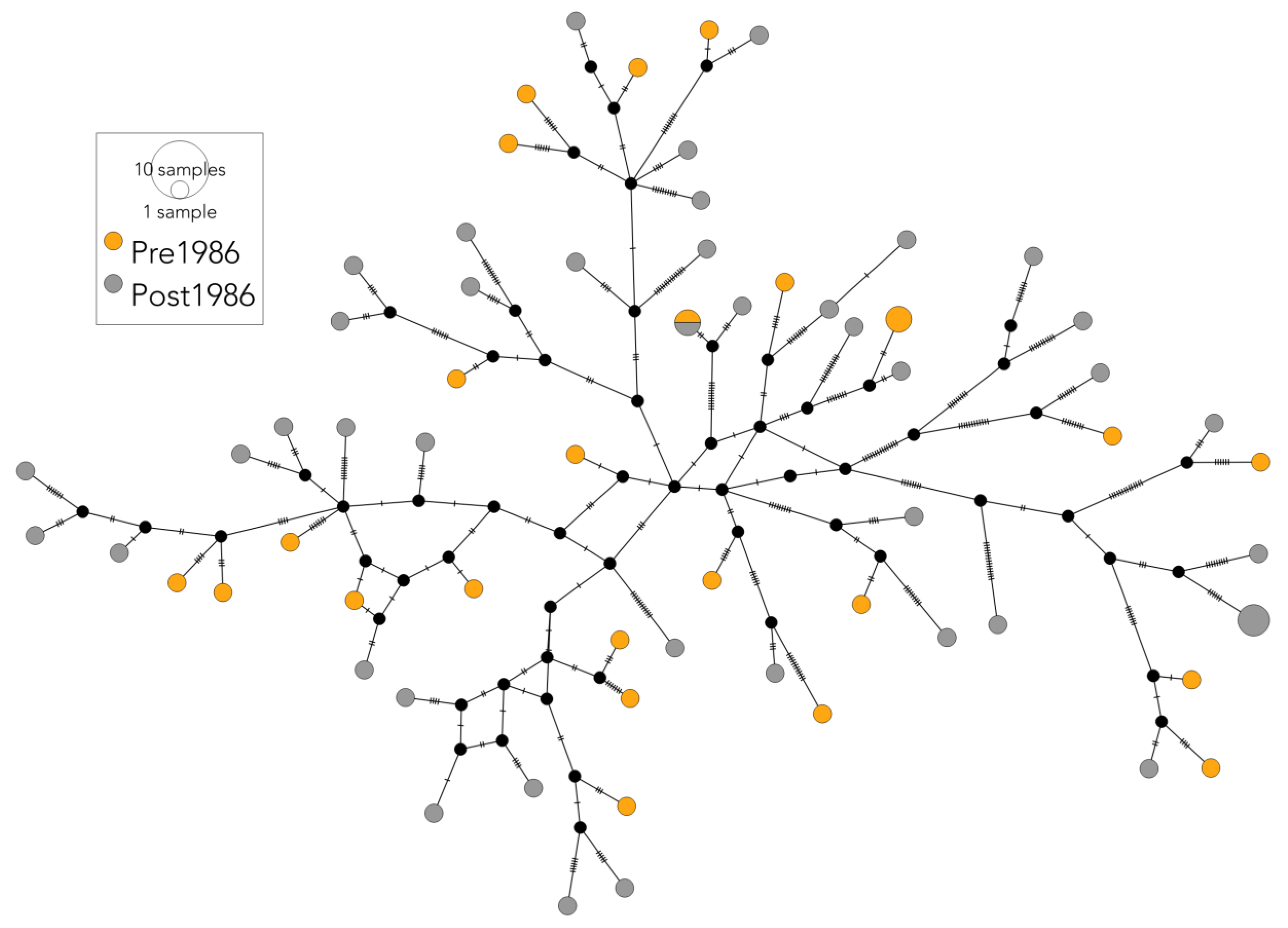
3.1. Mitogenome and mtDNA CR Sequencing of the Historic Fin Whale Samples
3.2. SHFW Genetic Diversity and Population Structure
3.2.1. Genetic Diversity
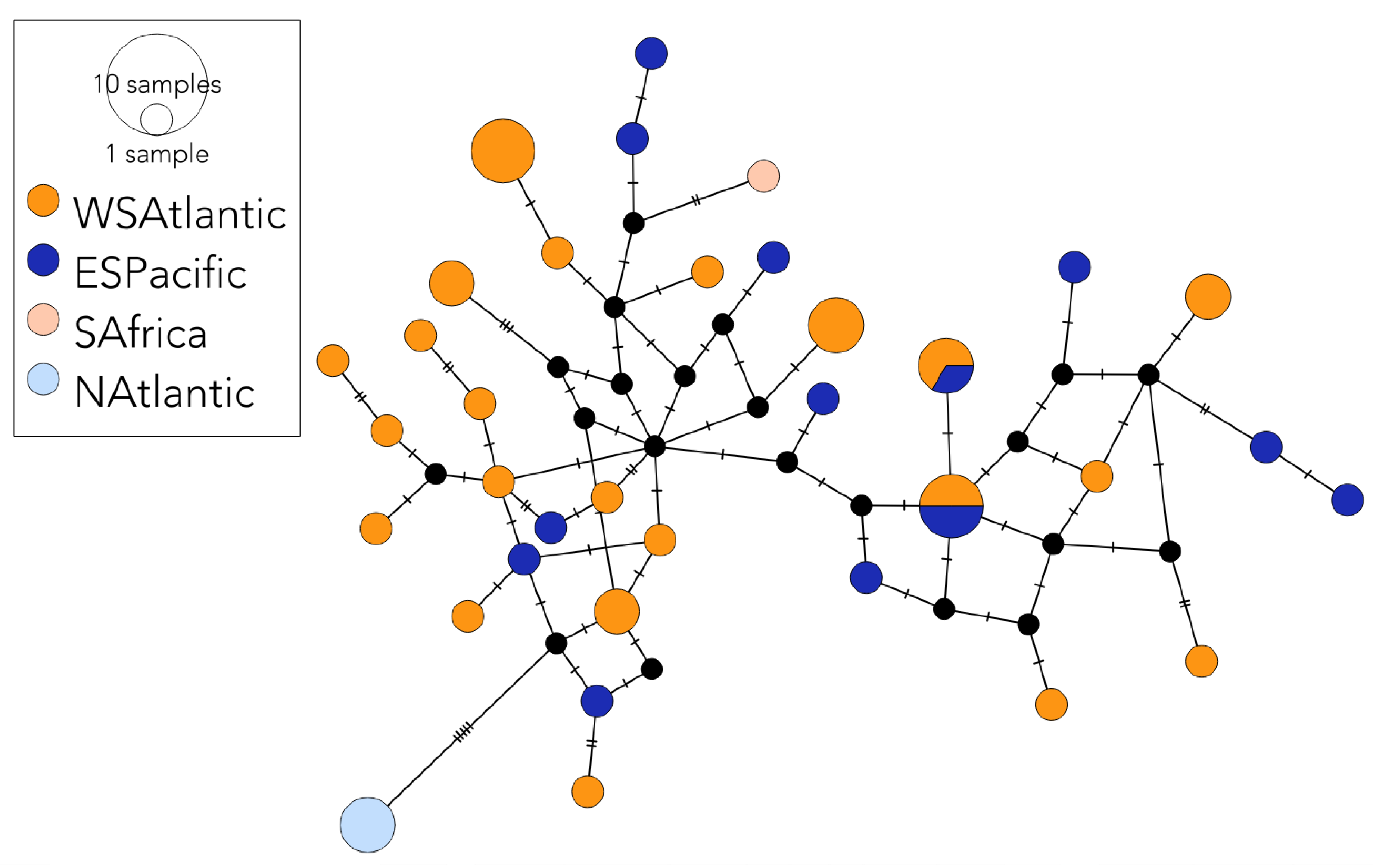
3.2.2. Population Structure
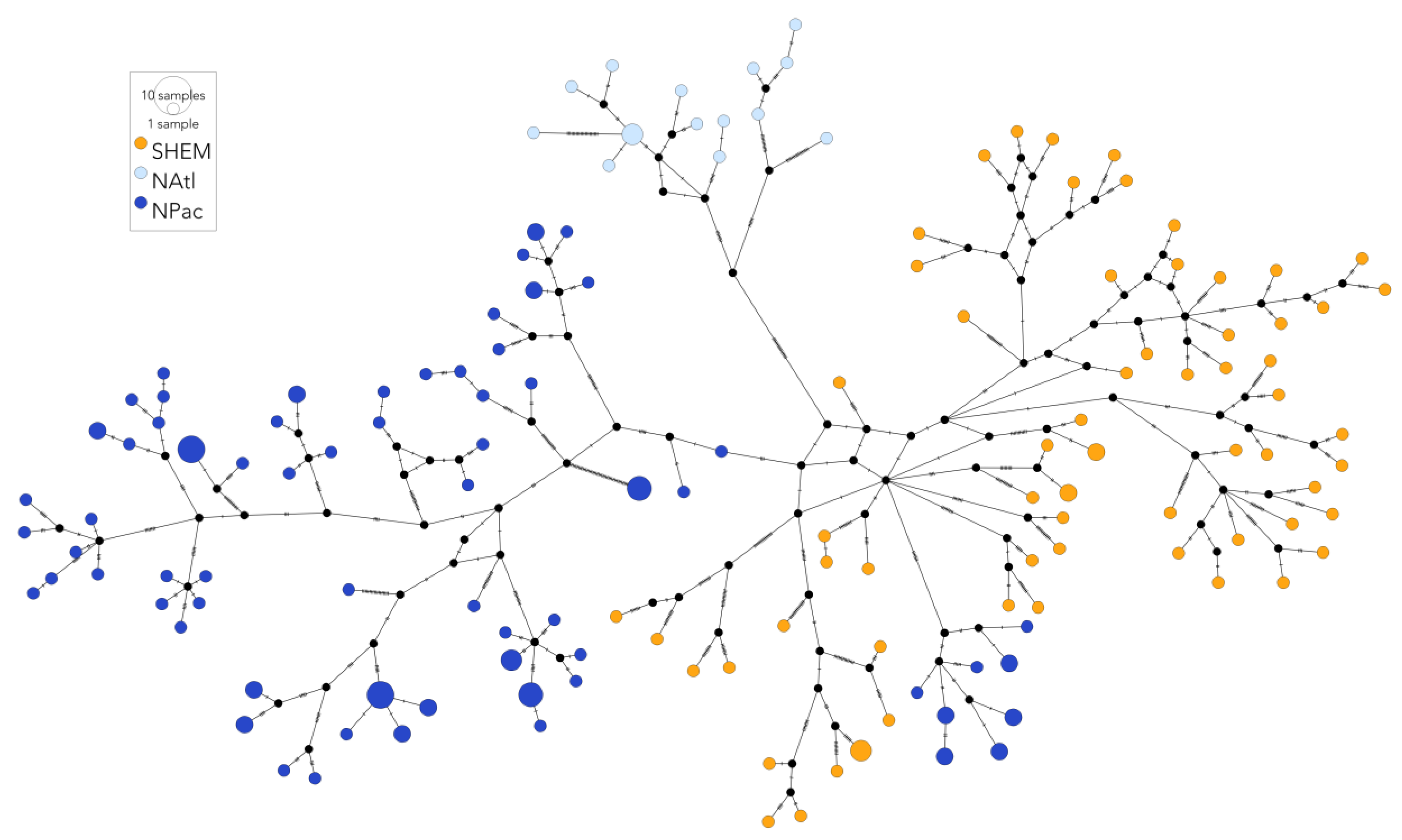
3.3. Comparison with Northern Hemisphere Fin Whales
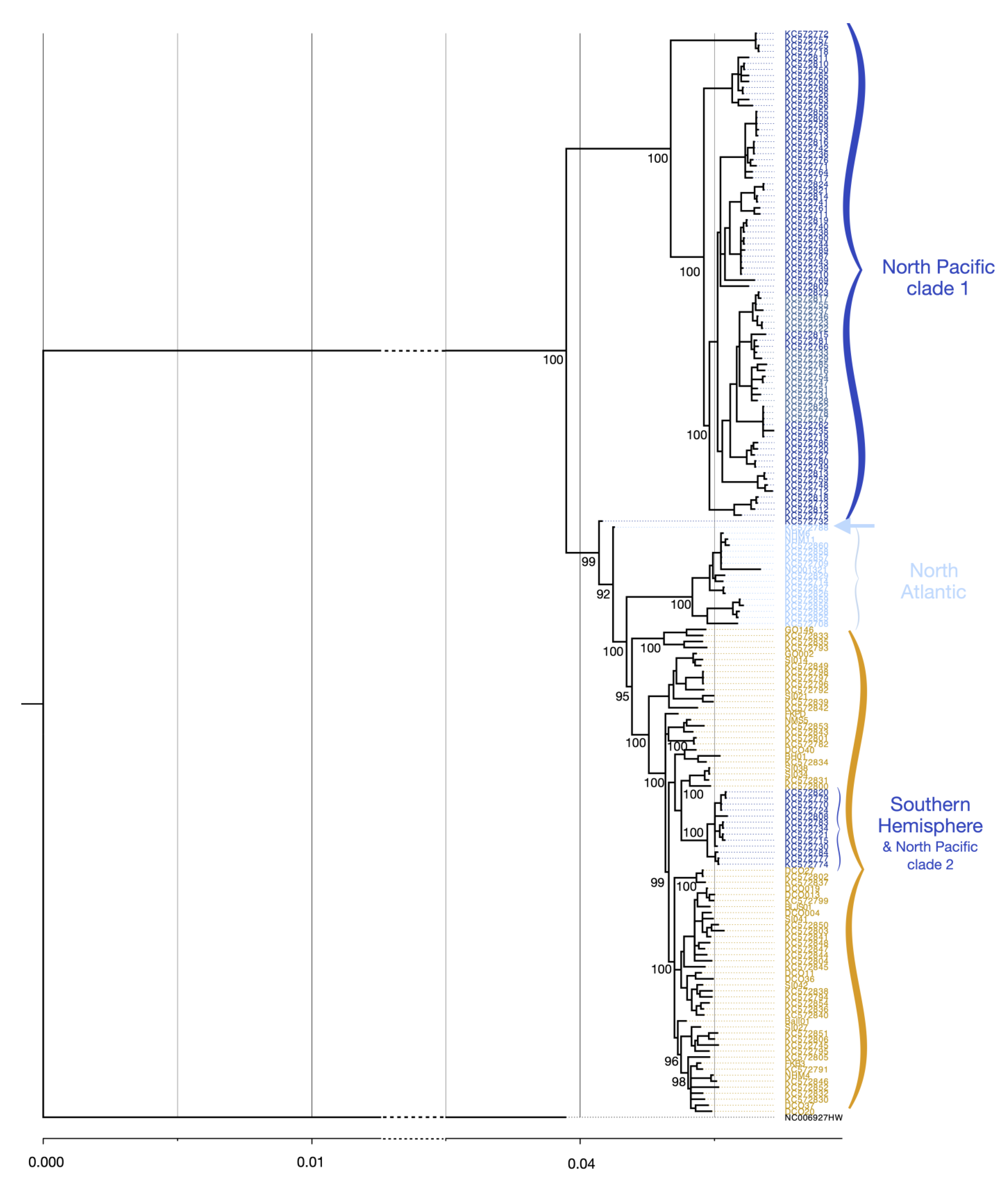
3.4. Mitogenome Phylogenetics
4. Discussion
4.1. Southern Hemisphere Population Structure
4.2. Global Population Structure
4.3. Fin Whale Recovery and Ecosystem Change
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roman, J.; Estes, J.A.; Morissette, L.; Smith, C.; Costa, D.; McCarthy, J.; Nation, J.B.; Nicol, S.; Pershing, A.; Smetacek, V. Whales as Marine Ecosystem Engineers. Front. Ecol. Environ. 2014, 12, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Willis, J. Whales Maintained a High Abundance of Krill; Both Are Ecosystem Engineers in the Southern Ocean. Mar. Ecol. Prog. Ser. 2014, 513, 51–69. [Google Scholar] [CrossRef]
- Smith, C.R.; Glover, A.G.; Treude, T.; Higgs, N.D.; Amon, D.J. Whale-Fall Ecosystems: Recent Insights into Ecology, Paleoecology, and Evolution. Ann. Rev. Mar. Sci. 2015, 7, 571–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizroch, S.A.; Rice, D.W. The Fin Whale, Balaenoptera physalus. Mar. Fish. Rev. 1984, 46, 20–24. [Google Scholar]
- Mori, M.; Butterworth, D.S. A First Step towards Modelling the Krill–predator Dynamics of the Antarctic Ecosystem. 2006. Available online: http://hdl.handle.net/11427/18458 (accessed on 1 July 2021).
- Savoca, M.S.; Czapanskiy, M.F.; Kahane-Rapport, S.R.; Gough, W.T.; Fahlbusch, J.A.; Bierlich, K.C.; Segre, P.S.; Di Clemente, J.; Penry, G.S.; Wiley, D.N.; et al. Baleen Whale Prey Consumption Based on High-Resolution Foraging Measurements. Nature 2021, 599, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Herr, H.; Viquerat, S.; Devas, F.; Lees, A.; Wells, L.; Gregory, B.; Giffords, T.; Beecham, D.; Meyer, B. Return of Large Fin Whale Feeding Aggregations to Historical Whaling Grounds in the Southern Ocean. Sci. Rep. 2022, 12, 9458. [Google Scholar] [CrossRef] [PubMed]
- Mackintosh, N.A. The Natural History of Whalebone Whales. Biol. Rev. Camb. Philos. Soc. 1946, 21, 60–74. [Google Scholar] [CrossRef]
- Mikhalev, Y.A.; Tormosov, D.D. Corrected Data about Non-Soviet Whale Marks Recovered by Soviet Whaling Fleets. Annu. Rep. Int. Whal. Comm. 1997, 47, 1019–1027. [Google Scholar]
- Mikhalev, Y. Whales of the Southern Ocean: Biology, Whaling and Perspectives of Population Recovery; Springer Nature: Cham, Switzerland, 2020; pp. 1–408. ISBN 9783030292522. [Google Scholar]
- Rocha, R.C., Jr.; Clapham, P.J.; Ivashchenko, Y. Emptying the Oceans: A Summary of Industrial Whaling Catches in the 20th Century. Mar. Fish. Rev. 2015, 76, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Allison, C. IWC Individual Whale Catch Database Version 6.1, 18 July 2016; IWC: Cambridge, UK, 2016; Available online: https://iwc.int/scientific-research/data-availability (accessed on 1 June 2020).
- Hohenlohe, P.A.; Funk, W.C.; Rajora, O.P. Population Genomics for Wildlife Conservation and Management. Mol. Ecol. 2021, 30, 62–82. [Google Scholar] [CrossRef]
- Archer, F.I.; Brownell, R.L.; Hancock-Hanser, B.L.; Morin, P.A.; Robertson, K.M.; Sherman, K.K.; Calambokidis, J.; Urbán, R.J.; Rosel, P.E.; Mizroch, S.A.; et al. Revision of Fin Whale Balaenoptera Physalus (Linnaeus, 1758) Subspecies Using Genetics. J. Mammal. 2019, 100, 1653–1670. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R. Pygmy Fin Whales. Mar. Mamm. Sci. 2004, 20, 329–334. [Google Scholar] [CrossRef]
- Pérez-Alvarez, M.; Kraft, S.; Segovia, N.I.; Olavarría, C.; Nigenda-Morales, S.; Urbán, R.J.; Viloria-Gómora, L.; Archer, F.; Moraga, R.; Sepúlveda, M.; et al. Contrasting Phylogeographic Patterns Among Northern and Southern Hemisphere Fin Whale Populations With New Data From the Southern Pacific. Front. Mar. Sci. 2021, 8. [Google Scholar] [CrossRef]
- Archer, F.I.; Morin, P.A.; Hancock-Hanser, B.L.; Robertson, K.M.; Leslie, M.S.; Bérubé, M.; Panigada, S.; Taylor, B.L. Mitogenomic Phylogenetics of Fin Whales (Balaenoptera Physalus Spp.): Genetic Evidence for Revision of Subspecies. PLoS ONE 2013, 8, e63396. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, A.A.; Hoekendijk, J.P.A.; Aguilar, A.; Barco, S.G.; Berrow, S.; Bloch, D.; Borrell, A.; Cunha, H.A.; Dalla Rosa, L.; Dias, C.P.; et al. Fin Whale (Balaenoptera physalus) Mitogenomics: A Cautionary Tale of Defining Sub-Species from Mitochondrial Sequence Monophyly. Mol. Phylogenet. Evol. 2019, 135, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Balcazar, N.E.; Tripovich, J.S.; Klinck, H.; Nieukirk, S.L.; Mellinger, D.K.; Dziak, R.P.; Rogers, T.L. Calls Reveal Population Structure of Blue Whales across the Southeast Indian Ocean and the Southwest Pacific Ocean. J. Mammal. 2015, 96, 1184–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, E.C.; Royer, J.-Y.; Alling, A.; Maslen, B.; Rogers, T.L. Multiple Pygmy Blue Whale Acoustic Populations in the Indian Ocean: Whale Song Identifies a Possible New Population. Sci. Rep. 2021, 11, 8762. [Google Scholar] [CrossRef]
- Schall, E.; Thomisch, K.; Boebel, O.; Gerlach, G.; Mangia Woods, S.; Roca, I.T.; Van Opzeeland, I. Humpback Whale Song Recordings Suggest Common Feeding Ground Occupation by Multiple Populations. Sci. Rep. 2021, 11, 18806. [Google Scholar] [CrossRef]
- Wood, M.; Širović, A. Characterization of Fin Whale Song off the Western Antarctic Peninsula. PLoS ONE 2022, 17, e0264214. [Google Scholar] [CrossRef]
- Rendell, L.; Cantor, M.; Gero, S.; Whitehead, H.; Mann, J. Causes and Consequences of Female Centrality in Cetacean Societies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180066. [Google Scholar] [CrossRef] [Green Version]
- Patenaude, N.J.; Portway, V.A.; Schaeff, C.M.; Bannister, J.L.; Best, P.B.; Payne, R.S.; Rowntree, V.J.; Rivarola, M.; Baker, C.S. Mitochondrial DNA Diversity and Population Structure among Southern Right Whales (Eubalaena australis). J. Hered. 2007, 98, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Baker, C.S.; Steel, D.; Calambokidis, J.; Falcone, E.; González-Peral, U.; Barlow, J.; Burdin, A.M.; Clapham, P.J.; Ford, J.K.B.; Gabriele, C.M.; et al. Strong Maternal Fidelity and Natal Philopatry Shape Genetic Structure in North Pacific Humpback Whales. Mar. Ecol. Prog. Ser. 2013, 494, 291–306. [Google Scholar] [CrossRef] [Green Version]
- Leroux, R.A.; Dutton, P.H.; Abreu-Grobois, F.A.; Lagueux, C.J.; Campbell, C.L.; Delcroix, E.; Chevalier, J.; Horrocks, J.A.; Hillis-Starr, Z.; Troëng, S.; et al. Re-Examination of Population Structure and Phylogeography of Hawksbill Turtles in the Wider Caribbean Using Longer mtDNA Sequences. J. Hered. 2012, 103, 806–820. [Google Scholar] [CrossRef] [Green Version]
- Shamblin, B.M.; Bolten, A.B.; Abreu-Grobois, F.A.; Bjorndal, K.A.; Cardona, L.; Carreras, C.; Clusa, M.; Monzón-Argüello, C.; Nairn, C.J.; Nielsen, J.T.; et al. Geographic Patterns of Genetic Variation in a Broadly Distributed Marine Vertebrate: New Insights into Loggerhead Turtle Stock Structure from Expanded Mitochondrial DNA Sequences. PLoS ONE 2014, 9, e85956. [Google Scholar] [CrossRef] [Green Version]
- Cammen, K.M.; Andrews, K.R.; Carroll, E.L.; Foote, A.D.; Humble, E.; Khudyakov, J.I.; Louis, M.; McGowen, M.R.; Olsen, M.T.; Van Cise, A.M. Genomic Methods Take the Plunge: Recent Advances in High-Throughput Sequencing of Marine Mammals. J. Hered. 2016, 107, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Rey-Iglesia, A.; Lister, A.M.; Campos, P.F.; Brace, S.; Mattiangeli, V.; Daly, K.G.; Teasdale, M.D.; Bradley, D.G.; Barnes, I.; Hansen, A.J. Exploring the Phylogeography and Population Dynamics of the Giant Deer (Megaloceros Giganteus) Using Late Quaternary Mitogenomes. Proc. Biol. Sci. 2021, 288, 20201864. [Google Scholar] [CrossRef]
- Shamblin, B.M.; Bjorndal, K.A.; Bolten, A.B.; Hillis-Starr, Z.M.; Lundgren, I.; Naro-Maciel, E.; Nairn, C.J. Mitogenomic Sequences Better Resolve Stock Structure of Southern Greater Caribbean Green Turtle Rookeries. Mol. Ecol. 2012, 21, 2330–2340. [Google Scholar] [CrossRef] [PubMed]
- Feutry, P.; Kyne, P.M.; Pillans, R.D.; Chen, X.; Marthick, J.; Morgan, D.L.; Grewe, P.M. Whole Mitogenome Sequencing Refines Population Structure of the Critically Endangered Sawfish Pristis Pristis. Mar. Ecol. Prog. Ser. 2015, 533, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Van Cise, A.M.; Baird, R.W.; Baker, C.S.; Cerchio, S.; Claridge, D.; Fielding, R.; Hancock-Hanser, B.; Marrero, J.; Martien, K.K.; Mignucci-Giannoni, A.A.; et al. Oceanographic Barriers, Divergence, and Admixture: Phylogeography and Taxonomy of Two Putative Subspecies of Short-Finned Pilot Whale. Mol. Ecol. 2019, 28, 2886–2902. [Google Scholar] [CrossRef] [PubMed]
- Kraft, D.W.; Conklin, E.E.; Barba, E.W.; Hutchinson, M.; Toonen, R.J.; Forsman, Z.H.; Bowen, B.W. Genomics versus mtDNA for Resolving Stock Structure in the Silky Shark (Carcharhinus falciformis). PeerJ 2020, 8, e10186. [Google Scholar] [CrossRef]
- Tønnessen, J.N.; Johnsen, A.O. The History of Modern Whaling; University of California Press: Berkeley, CA, USA, 1982; pp. 1–798. [Google Scholar]
- Taylor, B.L.; Chivers, S.J.; Larese, J.; Perrin, W.F. Generation Length and Percent Mature Estimates for IUCN Assessments of Cetaceans. 2007. Available online: https://citeseerx.ist.psu.edu/document?repid=rep1&type=pdf&doi=dd195ac71a51be8d92609ad7b888ea5d555eaea2 (accessed on 1 July 2022).
- Jackson, J.A.; Patenaude, N.J.; Carroll, E.L.; Baker, C.S. How Few Whales Were There after Whaling? Inference from Contemporary mtDNA Diversity. Mol. Ecol. 2008, 17, 236–251. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.A.; Carroll, E.L.; Smith, T.D.; Zerbini, A.N.; Patenaude, N.J.; Baker, C.S. An Integrated Approach to Historical Population Assessment of the Great Whales: Case of the New Zealand Southern Right Whale. R. Soc. Open Sci. 2016, 3, 150669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, E.L.; Alderman, R.; Bannister, J.L.; Bérubé, M.; Best, P.B.; Boren, L.; Baker, C.S.; Constantine, R.; Findlay, K.; Harcourt, R.; et al. Incorporating Non-Equilibrium Dynamics into Demographic History Inferences of a Migratory Marine Species. Heredity 2018, 122, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Von Thaden, A.; Cocchiararo, B.; Mueller, S.A.; Reiners, T.E.; Reinert, K.; Tuchscherer, I.; Janke, A.; Nowak, C. Informing Conservation Strategies with Museum Genomics: Long-Term Effects of Past Anthropogenic Persecution on the Elusive European Wildcat. Ecol. Evol. 2021, 11, 17932–17951. [Google Scholar] [CrossRef] [PubMed]
- Sremba, A.L.; Martin, A.R.; Scott Baker, C. Species Identification and Likely Catch Time Period of Whale Bones from South Georgia. Mar. Mamm. Sci. 2015, 31, 122–132. [Google Scholar] [CrossRef]
- Solazzo, C.; Fitzhugh, W.; Kaplan, S.; Potter, C.; Dyer, J.M. Molecular Markers in Keratins from Mysticeti Whales for Species Identification of Baleen in Museum and Archaeological Collections. PLoS ONE 2017, 12, e0183053. [Google Scholar] [CrossRef] [Green Version]
- Alter, S.E.; Rynes, E.; Palumbi, S.R. DNA Evidence for Historic Population Size and Past Ecosystem Impacts of Gray Whales. Proc. Natl. Acad. Sci. USA 2007, 104, 15162–15167. [Google Scholar] [CrossRef] [Green Version]
- Foote, A.D.; Hofreiter, M.; Morin, P.A. Ancient DNA from Marine Mammals: Studying Long-Lived Species over Ecological and Evolutionary Timescales. Ann. Anat. 2012, 194, 112–120. [Google Scholar] [CrossRef]
- Béland, S.L.; Frasier, B.A.; Darling, J.D.; Frasier, T.R. Using Pre- and Postexploitation Samples to Assess the Impact of Commercial Whaling on the Genetic Characteristics of Eastern North Pacific Gray and Humpback Whales and to Compare Methods Used to Infer Historic Demography. Mar. Mamm. Sci. 2020, 36, 398–420. [Google Scholar] [CrossRef]
- Sremba, A.L.; Martin, A.R.; Wilson, P.; Cypriano-Souze, A.L.; Buss, D.L.; Hart, T.; Engel, M.H.; Bonatto, S.L.; Rosenbaum, H.; Collins, T.; et al. Diversity of Mitochondrial DNA in Three Species of Great Whales before and after Modern Whaling. manuscript in preparation.
- Hutchison, D.W.; Templeton, A.R. Correlation of pairwise genetic and geographic distance measures: Inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 1999, 53, 1898–1914. [Google Scholar] [CrossRef]
- Colonna, V.; Pistis, G.; Bomba, L.; Mona, S.; Matullo, G.; Boano, R.; Sala, C.; Viganò, F.; Torroni, A.; Achilli, A.; et al. Small Effective Population Size and Genetic Homogeneity in the Val Borbera Isolate. Eur. J. Hum. Genet. 2013, 21, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Angst, P.; Ameline, C.; Haag, C.R.; Ben-Ami, F.; Ebert, D.; Fields, P.D. Genetic Drift Shapes the Evolution of a Highly Dynamic Metapopulation. Mol. Biol. Evol. 2022, 39. [Google Scholar] [CrossRef]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.-T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.-L.; et al. Complete Mitochondrial Genome Sequence of a Middle Pleistocene Cave Bear Reconstructed from Ultrashort DNA Fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [Green Version]
- Buss, D. Foraging Ecology and Population Structuring of Baleen Whales in the Western South Atlantic and Eastern South Pacific; University of Cambridge: Cambridge, UK, 2022; pp. 1–298. [Google Scholar] [CrossRef]
- Baker, C.S.; Medrano-Gonzalez, L.; Calambokidis, J.; Perry, A.; Pichler, F.; Rosenbaum, H.; Straley, J.M.; Urban-Ramirez, J.; Yamaguchi, M.; von Ziegesar, O. Population Structure of Nuclear and Mitochondrial DNA Variation among Humpback Whales in the North Pacific. Mol. Ecol. 1998, 7, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Garrigue, C.; Dodemont, R.; Steel, D.; Baker, C.S. Organismal And’gametic’capture-Recapture Using Microsatellite Genotyping Confirm Low Abundance and Reproductive Autonomy of Humpback Whales on the Wintering Grounds of New Caledonia. Mar. Ecol. Prog. Ser. 2004, 274, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Ross, H.A.; Lento, G.M.; Dalebout, M.L.; Goode, M.; Ewing, G.; McLaren, P.; Rodrigo, A.G.; Lavery, S.; Baker, C.S. DNA Surveillance: Web-Based Molecular Identification of Whales, Dolphins, and Porpoises. J. Hered. 2003, 94, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Kircher, M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harb. Protoc. 2010, 2010, db.prot5448. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid Adapter Trimming, Identification, and Read Merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Skoglund, P.; Northoff, B.H.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S.; Krause, J.; Jakobsson, M. Separating Endogenous Ancient DNA from Modern Day Contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. USA 2014, 111, 2229–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, M.; Ginolhac, A.; Lindgreen, S.; Thompson, J.F.; Al-Rasheid, K.A.S.; Willerslev, E.; Krogh, A.; Orlando, L. Improving Ancient DNA Read Mapping against Modern Reference Genomes. BMC Genom. 2012, 13, 178. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.W.; Bryant, D. Popart: Full-Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Alexander, A.; Steel, D.; Hoekzema, K.; Mesnick, S.L.; Engelhaupt, D.; Kerr, I.; Payne, R.; Baker, C.S. What Influences the Worldwide Genetic Structure of Sperm Whales (Physeter macrocephalus)? Mol. Ecol. 2016, 25, 2754–2772. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X. Statistical Tests of Neutrality of Mutations against Population Growth, Hitchhiking and Background Selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Arnason, U.; Gullberg, A.; Widegren, B. The Complete Nucleotide Sequence of the Mitochondrial DNA of the Fin Whale, Balaenoptera Physalus. J. Mol. Evol. 1991, 33, 556–568. [Google Scholar] [CrossRef]
- Sasaki, T.; Nikaido, M.; Hamilton, H.; Goto, M.; Kato, H.; Kanda, N.; Pastene, L.; Cao, Y.; Fordyce, R.; Hasegawa, M.; et al. Mitochondrial Phylogenetics and Evolution of Mysticete Whales. Syst. Biol. 2005, 54, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J. FigTree, Version 1.4. 0 2012. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 September 2022).
- Waples, R.S.; Hoelzel, A.R.; Gaggiotti, O. Guidelines for Genetic Data Analysis. J. Cetacean Res. Manag. 2018, 18, 33–80. [Google Scholar] [CrossRef]
- Baker, C.S.; Palumbi, S.R.; Lambertsen, R.H.; Weinrich, M.T.; Calambokidis, J.; O’Brien, S.J. Influence of Seasonal Migration on Geographic Distribution of Mitochondrial DNA Haplotypes in Humpback Whales. Nature 1990, 344, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Carroll, E.L.; Brooks, L.; Baker, C.S. Assessing the Design and Power of Capture-Recapture Studies to Estimate Demographic Parameters for the Endangered Oceania Humpback Whale Population. Endanger. Species Res. 2015, 28, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, H. Gene–culture Coevolution in Whales and Dolphins. Proc. Natl. Acad. Sci. USA 2017, 114, 7814–7821. [Google Scholar] [CrossRef] [Green Version]
- Frasier, B.A.; Springate, L.; Frasier, T.R.; Brewington, S.; Carruthers, M.; Edvardsson, R.; Harrison, R.; Kitchener, A.C.; Mainland, I.; Szabo, V.E. Genetic Examination of Historical North Atlantic Right Whale (Eubalaena glacialis) Bone Specimens from the Eastern North Atlantic: Insights into Species History, Transoceanic Population Structure, and Genetic Diversity. Mar. Mamm. Sci. 2022, 38, 1050–1069. [Google Scholar] [CrossRef]
- Schmitt, N.T.; Double, M.C.; Jarman, S.N.; Gales, N.; Marthick, J.R.; Polanowski, A.M.; Scott Baker, C.; Steel, D.; Jenner, K.C.S.; Jenner, M.-N.M.; et al. Low Levels of Genetic Differentiation Characterize Australian Humpback Whale (Megaptera novaeangliae) Populations. Mar. Mamm. Sci. 2014, 30, 221–241. [Google Scholar] [CrossRef]
- Carroll, E.L.; Ott, P.H.; Mcmillan, L.F.; Galletti Vernazzani, B.; Neveceralova, P.; Vermeulen, E.; Gaggiotti, O.E.; Andriolo, A.; Baker, C.S.; Bamford, C.; et al. Genetic Diversity and Connectivity of Southern Right Whales (Eubalaena australis) Found in the Chile-Peru Wintering Grounds and South Georgia (Islas georgias Del Sur) Feeding Grounds. Heredity 2020, 111, 263–276. [Google Scholar] [CrossRef]
- Olavarría, C.; Baker, C.S.; Garrigue, C.; Poole, M.; Hauser, N.; Caballero, S.; Flórez-González, L.; Brasseur, M.; Bannister, J.; Capella, J.; et al. Population Structure of South Pacific Humpback Whales and the Origin of the Eastern Polynesian Breeding Grounds. Mar. Ecol. Prog. Ser. 2007, 330, 257–268. [Google Scholar] [CrossRef]
- Hauser, N.; Zerbini, A.N.; Geyer, Y.; Heide-Jørgensen, M.-P.; Clapham, P. Movements of Satellite-Monitored Humpback Whales, Megaptera Novaeangliae, from the Cook Islands. Mar. Mamm. Sci. 2010, 26, 679–685. [Google Scholar] [CrossRef]
- Carvalho, I.; Loo, J.; Collins, T.; Barendse, J.; Pomilla, C.; Leslie, M.S.; Ngouessono, S.; Best, P.B.; Rosenbaum, H.C. Does Temporal and Spatial Segregation Explain the Complex Population Structure of Humpback Whales on the Coast of West Africa? Mar. Biol. 2014, 161, 805–819. [Google Scholar] [CrossRef] [Green Version]
- Waples, R.S.; Gaggiotti, O. What Is a Population? An Empirical Evaluation of Some Genetic Methods for Identifying the Number of Gene Pools and Their Degree of Connectivity. Mol. Ecol. 2006, 15, 1419–1439. [Google Scholar] [CrossRef] [PubMed]
- Foote, A.D.; Morin, P.A. Genome-Wide SNP Data Suggest Complex Ancestry of Sympatric North Pacific Killer Whale Ecotypes. Heredity 2016, 117, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Onoufriou, A.B.; Gaggiotti, O.E.; Aguilar de Soto, N.; McCarthy, M.L.; Morin, P.A.; Rosso, M.; Dalebout, M.; Davison, N.; Baird, R.W.; Baker, C.S.; et al. Biogeography in the Deep: Hierarchical Population Genomic Structure of Two Beaked Whale Species. Glob. Ecol. Conserv. 2022, 40, e02308. [Google Scholar] [CrossRef]
- Jackson, J.A.; Steel, D.J.; Beerli, P.; Congdon, B.C.; Olavarría, C.; Leslie, M.S.; Pomilla, C.; Rosenbaum, H.; Baker, C.S. Global Diversity and Oceanic Divergence of Humpback Whales (Megaptera novaeangliae). Proc. Biol. Sci. 2014, 281. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, H.C.; Kershaw, F.; Mendez, M.; Pomilla, C.; Leslie, M.S.; Findlay, K.P.; Best, P.B.; Collins, T.; Vely, M.; Engel, M.H.; et al. First Circumglobal Assessment of Southern Hemisphere Humpback Whale Mitochondrial Genetic Variation and Implications for Management. Endanger. Species Res. 2017, 32, 551–567. [Google Scholar] [CrossRef] [Green Version]
- Cypriano-Souza, A.L.; Engel, M.H.; Caballero, S.; Olavarría, C.; Flórez-González, L.; Capella, J.; Steel, D.; Sremba, A.; Aguayo, A.; Thiele, D.; et al. Genetic Differentiation between Humpback Whales ( Megaptera Novaeangliae ) from Atlantic and Pacific Breeding Grounds of South America. Mar. Mamm. Sci. 2017, 33, 457–479. [Google Scholar] [CrossRef]
- Sremba, A.L.; Hancock-Hanser, B.; Branch, T.A.; LeDuc, R.L.; Baker, C.S. Circumpolar Diversity and Geographic Differentiation of mtDNA in the Critically Endangered Antarctic Blue Whale (Balaenoptera Musculus Intermedia). PLoS ONE 2012, 7, e32579. [Google Scholar] [CrossRef] [Green Version]
- Attard, C.R.M.; Beheregaray, L.B.; Möller, L.M. Towards Population-Level Conservation in the Critically Endangered Antarctic Blue Whale: The Number and Distribution of Their Populations. Sci. Rep. 2016, 6, 22291. [Google Scholar] [CrossRef] [Green Version]
- Branch, T.A.; Stafford, K.M.; Palacios, D.M.; Allison, C.; Bannister, J.L.; Burton, C.L.K.; Cabrera, E.; Carlson, C.A.; Galletti Vernazzani, B.; Gill, P.C.; et al. Past and Present Distribution, Densities and Movements of Blue Whales Balaenoptera Musculus in the Southern Hemisphere and Northern Indian Ocean. Mamm. Rev. 2007, 37, 116–175. [Google Scholar] [CrossRef] [Green Version]
- Andrews-Goff, V.; Bell, E.M.; Miller, B.S.; Wotherspoon, S.J.; Double, M.C. Satellite Tag Derived Data from Two Antarctic Blue Whales (Balaenopteramusculusintermedia) Tagged in the East Antarctic Sector of the Southern Ocean. Biodivers. Data J. 2022, 10, e94228. [Google Scholar] [CrossRef] [PubMed]
- Rand, Z.; Branch, T.; Jackson, J.A. High Movement Rates of Antarctic Blue Whales (Balaenoptera Musculus Intermedia) on Southern Ocean Feeding Grounds Estimated from Historic Discovery Marks. manuscript in preparation.
- Sepúlveda, M.; Pérez-Álvarez, M.J. From Whaling to Whale Watching: Identifying Fin Whale Critical Foraging Habitats off the Chilean Coast. Aquatic 2018, 28, 821–829. [Google Scholar] [CrossRef]
- Buss, D.L.; Hearne, E.; Loy, R.H.Y.; Manica, A.; O’Connell, T.C.; Jackson, J.A. Evidence of Resource Partitioning between Fin and Sei Whales during the Twentieth-Century Whaling Period. Mar. Biol. 2022, 169. [Google Scholar] [CrossRef]
- Shabangu, F.W.; Findlay, K.P.; Yemane, D.; Stafford, K.M.; van den Berg, M.; Blows, B.; Andrew, R.K. Seasonal Occurrence and Diel Calling Behaviour of Antarctic Blue Whales and Fin Whales in Relation to Environmental Conditions off the West Coast of South Africa. J. Mar. Syst. 2019, 190, 25–39. [Google Scholar] [CrossRef]
- Constaratas, A.N.; McDonald, M.A.; Goetz, K.T.; Giorli, G. Fin Whale Acoustic Populations Present in New Zealand Waters: Description of Song Types, Occurrence and Seasonality Using Passive Acoustic Monitoring. PLoS ONE 2021, 16, e0253737. [Google Scholar] [CrossRef]
- Edwards, E.F.; Hall, C.; Moore, T.J.; Sheredy, C.; Redfern, J.V. Global Distribution of Fin Whales B Alaenoptera Physalus in the Post-Whaling Era (1980–2012). Mamm. Rev. 2015, 45, 197–214. [Google Scholar] [CrossRef]
- Aguilar, A.; García-Vernet, R. Fin Whale: Balaenoptera Physalus. In Encyclopedia of Marine Mammals, 3rd ed.; Würsig, B., Thewissen, J.G.M., Kovacs, K.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 368–371. ISBN 9780128043271. [Google Scholar]
- Lyrholm, T.; Leimar, O.; Johanneson, B.; Gyllensten, U. Sex–biased Dispersal in Sperm Whales: Contrasting Mitochondrial and Nuclear Genetic Structure of Global Populations. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1999, 266, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foote, A.D.; Vilstrup, J.T.; De Stephanis, R.; Verborgh, P.; Abel Nielsen, S.C.; Deaville, R.; Kleivane, L.; Martín, V.; Miller, P.J.O.; Oien, N.; et al. Genetic Differentiation among North Atlantic Killer Whale Populations. Mol. Ecol. 2011, 20, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Leduc, R.G.; Archer, F.I.; Lang, A.R.; Martien, K.K.; Hancock-Hanser, B.; Torres-Florez, J.P.; Hucke-Gaete, R.; Rosenbaum, H.C.; van Waerebeek, K.; Brownell, R.L., Jr.; et al. Genetic Variation in Blue Whales in the Eastern Pacific: Implication for Taxonomy and Use of Common Wintering Grounds. Mol. Ecol. 2017, 26, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.S.; Perry, A.; Bannister, J.L.; Weinrich, M.T.; Abernethy, R.B.; Calambokidis, J.; Lien, J.; Lambertsen, R.H.; Ramírez, J.U.; Vasquez, O. Abundant Mitochondrial DNA Variation and World-Wide Population Structure in Humpback Whales. Proc. Natl. Acad. Sci. USA 1993, 90, 8239–8243. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, H.C.; Brownell, R.L.; Brown, M.W.; Schaeff, C.; Portway, V.; White, B.N.; Malik, S.; Pastene, L.A.; Patenaude, N.J.; Baker, C.S.; et al. World-Wide Genetic Differentiation of Eubalaena: Questioning the Number of Right Whale Species. Mol. Ecol. 2000, 9, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Gaines, C.A.; Hare, M.P.; Beck, S.E.; Rosenbaum, H.C. Nuclear Markers Confirm Taxonomic Status and Relationships among Highly Endangered and Closely Related Right Whale Species. Proc. Biol. Sci. 2005, 272, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Huijser, L.A.E.; Bérubé, M.; Cabrera, A.A.; Prieto, R.; Silva, M.A.; Robbins, J.; Kanda, N.; Pastene, L.A.; Goto, M.; Yoshida, H.; et al. Population Structure of North Atlantic and North Pacific Sei Whales (Balaenoptera borealis) Inferred from Mitochondrial Control Region DNA Sequences and Microsatellite Genotypes. Conserv. Genet. 2018, 19, 1007–1024. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Álvarez, M.J.; Rodríguez, F.; Kraft, S.; Segovia, N.; Olavarría, C.; Baker, C.S.; Steel, D.; Funahashi, N.; Häussermann, V.; Ulloa, M.; et al. Phylogeography and Demographic Inference of the Endangered Sei Whale, with Implications for Conservation. Aquat. Conserv. 2021. [Google Scholar] [CrossRef]
- Cabrera, A.A.; Schall, E.; Bérubé, M.; Anderwald, P.; Bachmann, L.; Berrow, S.; Best, P.B.; Clapham, P.J.; Cunha, H.A.; Dalla Rosa, L.; et al. Strong and Lasting Impacts of Past Global Warming on Baleen Whales and Their Prey. Glob. Change Biol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.G. The Movements of Fin and Blue Whales in Antarctic Waters. Discovery. Reports. 1962, 33, 1–54. Available online: https://www.biodiversitylibrary.org/bibliography/6168 (accessed on 1 July 2021).
- Herr, H.; Viquerat, S.; Siegel, V.; Kock, K.-H.; Dorschel, B.; Huneke, W.G.C.; Bracher, A.; Schröder, M.; Gutt, J. Horizontal Niche Partitioning of Humpback and Fin Whales around the West Antarctic Peninsula: Evidence from a Concurrent Whale and Krill Survey. Polar Biol. 2016, 39, 799–818. [Google Scholar] [CrossRef]
- Viquerat, S.; Herr, H. Mid-Summer Abundance Estimates of Fin Whales Balaenoptera Physalus around the South Orkney Islands and Elephant Island. Endanger. Species Res. 2017, 32, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Barendse, J.; Best, P.B.; Carvalho, I.; Pomilla, C. Mother Knows Best: Occurrence and Associations of Resighted Humpback Whales Suggest Maternally Derived Fidelity to a Southern Hemisphere Coastal Feeding Ground. PLoS ONE 2013, 8, e81238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, A.R.; Loo, J.; Jaris, H.; Olavarria, C.; Thiele, D.; Ensor, P.; Aguayo, A.; Rosenbaum, H.C. Population Genetic Structure among Feeding Aggregations of Humpback Whales in the Southern Ocean. Mar. Biol. 2016, 163, 132. [Google Scholar] [CrossRef]
- Cade, D.E.; Friedlaender, A.S.; Calambokidis, J.; Goldbogen, J.A. Kinematic Diversity in Rorqual Whale Feeding Mechanisms. Curr. Biol. 2016, 26, 2617–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hückstädt, L.A.; Burns, J.M.; Koch, P.L.; McDonald, B.I.; Crocker, D.E.; Costa, D.P. Diet of a Specialist in a Changing Environment: The Crabeater Seal along the Western Antarctic Peninsula. Mar. Ecol. Prog. Ser. 2012, 455, 287–301. [Google Scholar] [CrossRef] [Green Version]
- McMahon, K.W.; Michelson, C.I.; Hart, T.; McCarthy, M.D.; Patterson, W.P.; Polito, M.J. Divergent Trophic Responses of Sympatric Penguin Species to Historic Anthropogenic Exploitation and Recent Climate Change. Proc. Natl. Acad. Sci. USA 2019, 116, 25721–25727. [Google Scholar] [CrossRef]
- Viquerat, S.; Waluda, C.M.; Kennedy, A.S.; Jackson, J.A.; Hevia, M.; Carroll, E.L.; Buss, D.L.; Burkhardt, E.; Thain, S.; Smith, P. Secchi, E.R. Identifying Seasonal Distribution Patterns of Fin Whales across the Scotia Sea and the Antarctic Peninsula Region Using a Novel Approach Combining Habitat Suitability Models and Ensemble Learning Methods. Front. Mar. Sci. 2022, 9. [Google Scholar] [CrossRef]
- Trathan, P.N.; Forcada, J.; Murphy, E.J. Environmental Forcing and Southern Ocean Marine Predator Populations: Effects of Climate Change and Variability. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 2351–2365. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, A.; Hill, S.L.; Pakhomov, E.A.; Siegel, V.; Reiss, C.S.; Loeb, V.J.; Steinberg, D.K.; Schmidt, K.; Tarling, G.A.; Gerrish, L.; et al. Krill (Euphausia superba) Distribution Contracts Southward during Rapid Regional Warming. Nat. Clim. Change 2019, 9, 142–147. [Google Scholar] [CrossRef]
- Bestley, S.; Ropert-Coudert, Y.; Bengtson Nash, S.; Brooks, C.M.; Cotté, C.; Dewar, M.; Friedlaender, A.S.; Jackson, J.A.; Labrousse, S.; Lowther, A.D.; et al. Marine Ecosystem Assessment for the Southern Ocean: Birds and Marine Mammals in a Changing Climate. Front. Ecol. Evol. 2020, 8, 338. [Google Scholar] [CrossRef]
- Širović, A.; Oleson, E.M. The Bioacoustics of Blue Whales—Global Diversity and Behavioral Variability in a Foraging Specialist. In Ethology and Behavioral Ecology of Mysticetes; Clark, C.W., Garland, E.C., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 195–221. ISBN 9783030984496. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population [source] | No. of Samples | No. of Unique Haplotypes | Polymorphic Sites | Hd (sd) | Nd (sd) | Td [p] | Fu’s F [p] |
|---|---|---|---|---|---|---|---|
| SHFWs, historical | |||||||
| Eastern South Pacific (SPhist) [This study] | 11 | 11 | 86 {32} | 1.0 (0.04) | 0.0014 (0.0007) | −1.39 [0.07] | −2.17 [0.08] |
| Western South Atlantic (SAhist) [This study] | 13 | 12 | 155 {37} | 0.987 (0.04) | 0.002 (0.0011) | −1.70 [0.04] | −2.09 [0.10] |
| Circumpolar [This study] | 25 | 24 | 222 {66} | 0.99 (0.01) | 0.002 (0.000) | −2.14 [0.00] | −8.69 [0.00] |
| SHFWs, contemporary | |||||||
| Eastern South Atlantic (SAe) [17] | 38 | 36 | 329 {185} | 0.996 (0.008) | 0.002 (0.0010) | −2.24 [0.00] | −16.55 [0.00] |
| Circumpolar [17] | 43 | 41 | 362 {206} | 0.99 (0.01) | 0.002 (0.000) | −2.30 [0.00] | −20.6 [0.00] |
| Population [source] | No. of Samples | No. of Unique Haplotypes | Polymorphic Sites | Hd (sd) | Nd (sd) | Td [p] | Fu’s F [p] |
|---|---|---|---|---|---|---|---|
| SHFWs, historical | |||||||
| Eastern South Pacific (SPhist) [This study] | 14 | 14 | 11 {1} | 1.0 (0.03) | 0.015 (0.009) | −0.21 [0.50] | −13.9 [0.00] |
| Western South Atlantic (SAhist) [This study] | 32 | 32 | 17 {1} | 1.0 (0.008) | 0.016 (0.009) | −0.55 [0.92] | −26.0 [0.00] |
| Circumpolar [This study] | 47 | 32 | 20 {2} | 0.976 (0.011) | 0.016 (0.009) | −0.74 [0.26] | −26.1 [0.00] |
| SHFWs, contemporary | |||||||
| Eastern South Atlantic (SAe) [17] | 46 | 46 | 25 {4} | 1.0 (0.005) | 0.016 (0.009) | −1.26 [0.11] | −26.1 [0.00] |
| Eastern South Pacific (Ant.Pen) (SPe) [18] | 18 | 18 | 17 {2} | 1.0 (0.018) | 0.016 (0.009) | −1.14 [0.12] | −20.1 [0.00] |
| Eastern South Pacific (Chile) (SPchile) [16,17] | 37 | 37 | 16 {3} | 1.0 (0.006) | 0.015 (0.008) | −0.40 [0.38] | −26.1 [0.00] |
| Circumpolar [16,17,18] | 102 | 54 | 30 {12} | 0.978 (0.006) | 0.016 (0.008) | −1.20 [0.09] | −26.2 [0.00] |
| Eastern South Pacific pre-1986 (SPhist) | Western South Atlantic pre-1986 (SAhist) | Eastern South Atlantic post-1986 (SAe) | Eastern South Pacific (Antarctic Peninsula) post-1986 (SPe) | Eastern South Pacific (Chile) post-1986 (SPchile) | |
| Eastern South Pacific pre-1986 (SPhist) | 0.03 (<0.05) | 0.01 (<0.05) | 0.05 (<0.01) | 0.03 (<0.01) | |
| Western South Atlantic pre-1986 (SAhist) | 0.004 (0.35) | 0.03 (<0.001) | 0.06 (<0.001) | 0.04 (<0.001) | |
| Eastern South Atlantic post-1986 (SAe) | −0.016 (0.78) | −0.00 (0.45) | 0.05 (<0.001) | 0.03 (<0.001) | |
| Eastern South Pacific (Antarctic Peninsula) post-1986 (SPe) | −0.008 (0.56) | −0.015 (0.77) | −0.01 (0.55) | 0.06 (<0.001) | |
| Eastern South Pacific (Chile) post-1986 (SPchile) | −0.015 (0.70) | 0.001 (0.25) | −0.00 (0.43) | 0.02 (0.17) |
| Dataset [source] | No. of Samples | Unique Haps | P. Sites | Hd (sd) | Nd (sd) | Td [p] | Fu’s F [p] |
|---|---|---|---|---|---|---|---|
| Mito (no model of sequence evolution) | |||||||
| North Atlantic [17], this study | 16 | 14 | 87 {64} | 0.975 (0.004) | 0.0015 (0.0008) | −0.76 [0.22] | −1.29 [0.024] |
| North Pacific [17] | 96 | 67 | 326 {251} | 1.0 (0.002) | 0.0024 (0.0012) | −1.57 [0.027] | −12.78 [0.00] |
| Southern Hemisphere circumpolar [17], this study | 68 | 64 | 428 {348} | 0.998 (0.003) | 0.0021 (0.0010) | −2.34 [0.00] | −24.08 [0.00] |
| mtDNA CR (model of sequence evolution: Tamura and Nei—gamma = 0.42) | |||||||
| NA—without Mediterranean [17,18], this study | 355 | 72 | 39 {0} | 0.903 (0.009) | 0.019 (0.010) | −0.88 [0.21] | −25.3 [0.00] |
| NA—Mediterranean only [17,18] | 108 | 16 | 19 {0} | 0.784 (0.027) | 0.012 (0.007) | −0.85 [0.22] | −26.8 [0.00] |
| NP—without Gulf of California [16,17,18] | 346 | 33 | 25 {0} | 0.804 (0.015) | 0.008 (0.005) | −1.45 [0.05] | −27.5 [0.00] |
| NP—Gulf of California only [16,17,18] | 521 | 8 | 7 {3} | 0.16 (0.02) | 0.001 (0.001) | −1.58 [0.01] | −9.04 [0.00] |
| South Pacific [This study] | 70 | 41 | 25 {6} | 0.975 (0.008) | 0.016 (0.008) | −1.07 [0.14] | −26.2 [0.00] |
| South Atlantic [This study] | 79 | 46 | 26 {7} | 0.977 (0.006) | 0.016 (0.009) | −1.00 [0.15] | −26.1 [0.00] |
| Circumpolar [16,17,18], this study | 149 | 69 | 32 {4} | 0.979 (0.004) | 0.016 (0.008) | −1.16 [0.13] | −26.0 [0.00] |
| North Atlantic (NA) | North Pacific (NP) | Southern Hemisphere (SH) | |
| North Atlantic (NA) | 0.29 (<0.001) | 0.28 (<0.001) | |
| North Pacific (NP) | 0.46 (<0.001) | 0.53 (<0.001) | |
| Southern Hemisphere (SH) | 0.44 (<0.001) | 0.27 (<0.001) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buss, D.L.; Atmore, L.M.; Zicos, M.H.; Goodall-Copestake, W.P.; Brace, S.; Archer, F.I.; Baker, C.S.; Barnes, I.; Carroll, E.L.; Hart, T.; et al. Historical Mitogenomic Diversity and Population Structuring of Southern Hemisphere Fin Whales. Genes 2023, 14, 1038. https://doi.org/10.3390/genes14051038
Buss DL, Atmore LM, Zicos MH, Goodall-Copestake WP, Brace S, Archer FI, Baker CS, Barnes I, Carroll EL, Hart T, et al. Historical Mitogenomic Diversity and Population Structuring of Southern Hemisphere Fin Whales. Genes. 2023; 14(5):1038. https://doi.org/10.3390/genes14051038
Chicago/Turabian StyleBuss, Danielle L., Lane M. Atmore, Maria H. Zicos, William P. Goodall-Copestake, Selina Brace, Frederick I. Archer, C. Scott Baker, Ian Barnes, Emma L. Carroll, Tom Hart, and et al. 2023. "Historical Mitogenomic Diversity and Population Structuring of Southern Hemisphere Fin Whales" Genes 14, no. 5: 1038. https://doi.org/10.3390/genes14051038
APA StyleBuss, D. L., Atmore, L. M., Zicos, M. H., Goodall-Copestake, W. P., Brace, S., Archer, F. I., Baker, C. S., Barnes, I., Carroll, E. L., Hart, T., Kitchener, A. C., Sabin, R., Sremba, A. L., Weir, C. R., & Jackson, J. A. (2023). Historical Mitogenomic Diversity and Population Structuring of Southern Hemisphere Fin Whales. Genes, 14(5), 1038. https://doi.org/10.3390/genes14051038