
Single-Locus and Multi-Locus Genome-Wide Association Studies Identify Genes Associated with Liver Cu Concentration in Merinoland Sheep

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample and Data Collection
2.2. Determination of Liver Copper Concentration
2.3. Genotyping and Quality Control
2.4. Single-Locus and Multi-Locus Genome-Wide Association Analyses
2.5. Gene Annotation and Enrichment
3. Results
3.1. Liver Copper Levels and Estimated Heritability
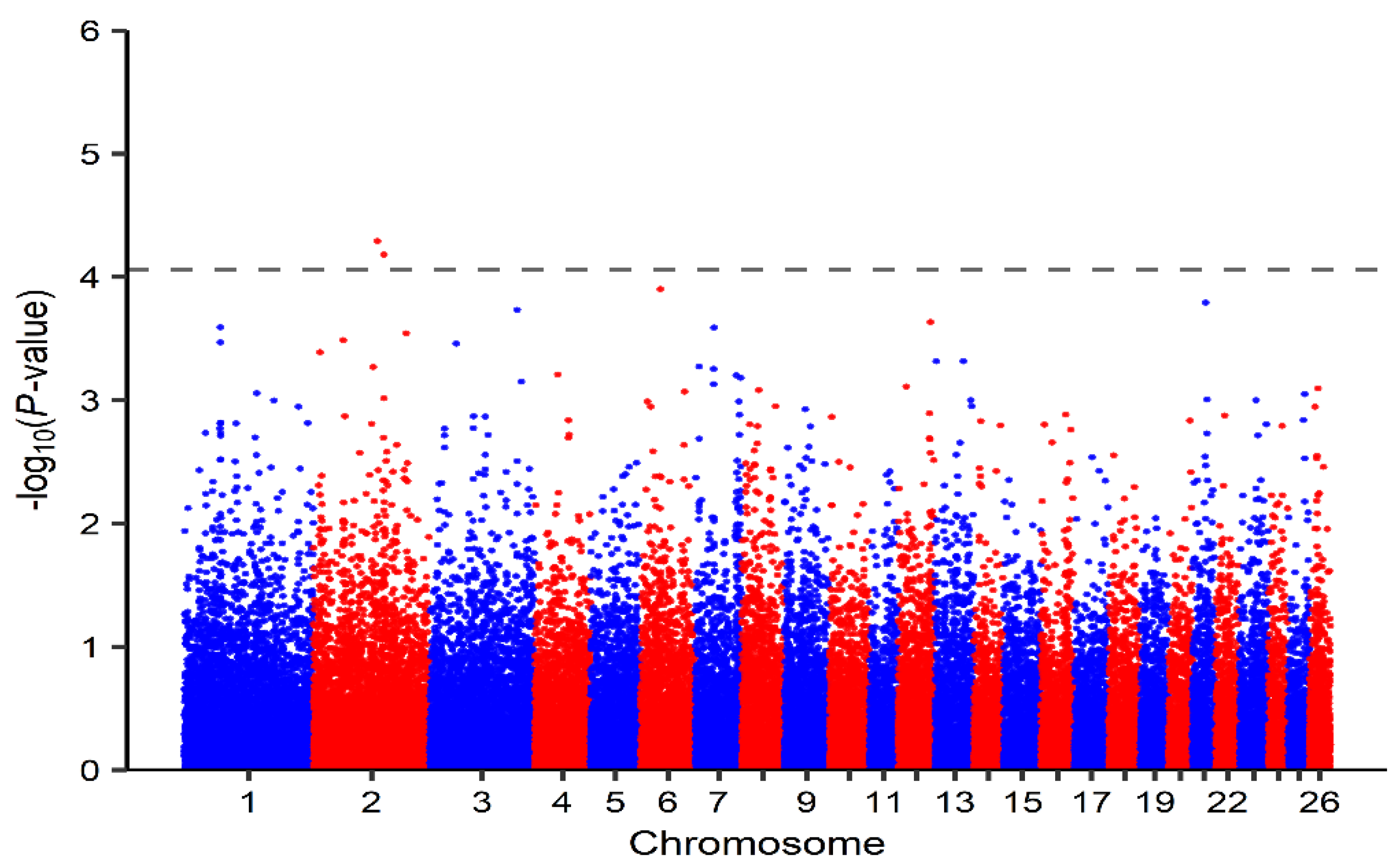
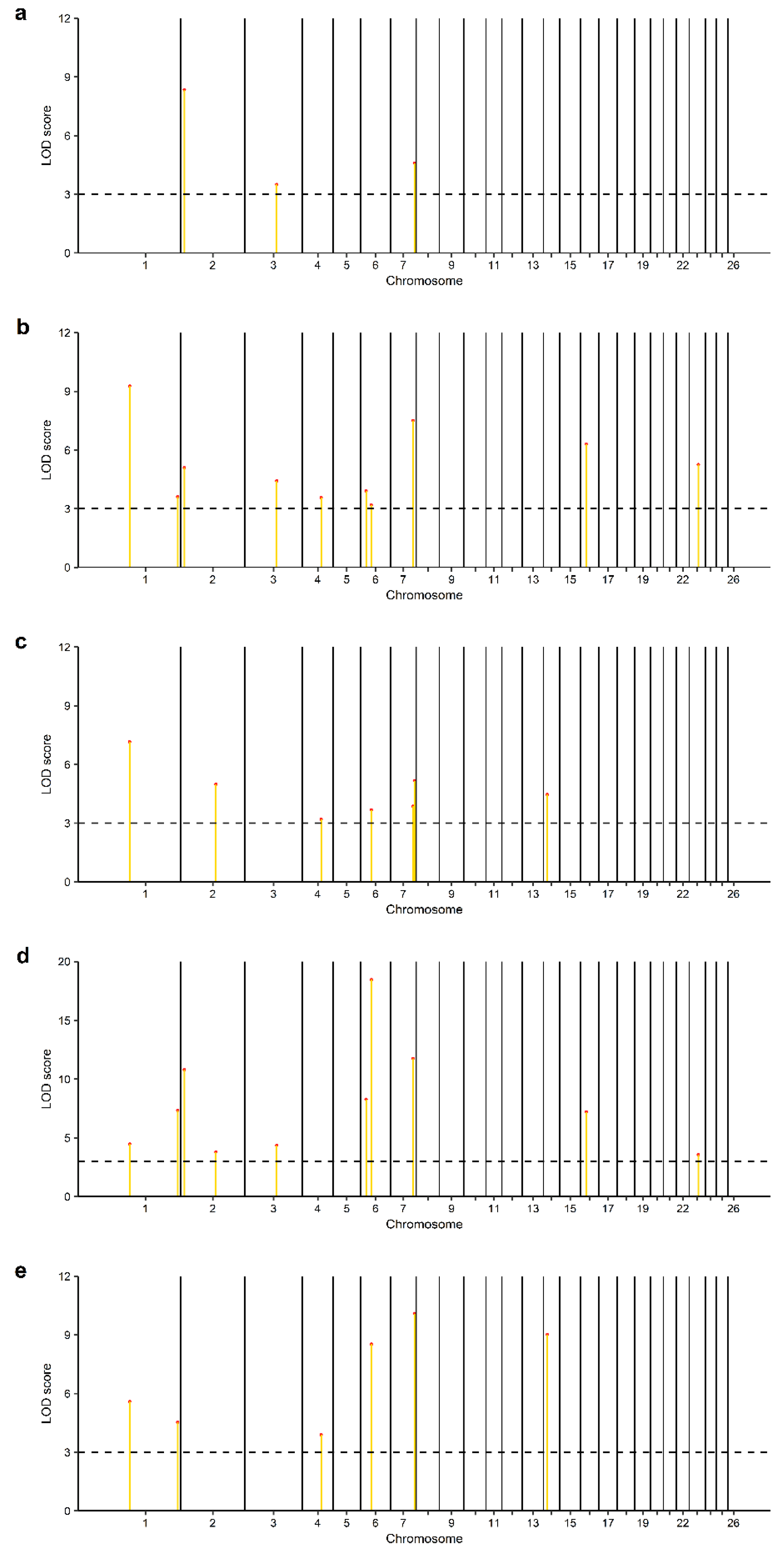
3.2. Identification of Genomic Regions and Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suttle, N.F. Mineral Nutrition of Livestock, 4th ed.; CABI: Cambridge, UK, 2010; ISBN 978-1-84593-472-9. [Google Scholar]
- De Bie, P.; Van De Sluis, B.; Klomp, L.; Wijmenga, C. The many faces of the copper metabolism protein MURR1/COMMD1. J. Hered. 2005, 96, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Haywood, S.; Simpson, D.M.; Ross, G.; Beynon, R.J. The greater susceptibility of North Ronaldsay sheep compared with Cambridge sheep to copper-induced oxidative stress, mitochondrial damage and hepatic stellate cell activation. J. Comp. Pathol. 2005, 133, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Suttle, N.F.; Jones, D.G. Micronutrient imbalance. In Diseases of Sheep, 4th ed.; Aitken, I.D., Ed.; Blackwell: Oxford, UK, 2007; pp. 377–394. [Google Scholar]
- Underwood, E.J.; Suttle, N.F. The Mineral Nutrition of Livestock, 3rd ed.; CABI: Cambridge, UK, 1999. [Google Scholar]
- Puls, R. Mineral Levels in Animal Health: Diagnostic Data; Sherpa International: Clearbrook, BC, Canada, 1994; ISBN 9780969342922. [Google Scholar]
- Kumaratilake, J.S. Chronic Copper Poisoning in Sheep: Liver Injury. JTEA 2014, 3, 1–22. [Google Scholar] [CrossRef]
- Sousa, I.K.F.d.; Hamad Minervino, A.H.; Sousa, R.D.S.; Chaves, D.F.; Soares, H.S.; Barros, I.d.O.; Araújo, C.A.S.C.d.; Júnior, R.A.B.; Ortolani, E.L. Copper deficiency in sheep with high liver iron accumulation. Vet. Med. Int. 2012, 2012, 207950. [Google Scholar] [CrossRef]
- Suttle, N.F.; Lewis, R.M.; Small, J.N.W. Effects of breed and family on rate of copper accretion in the liver of purebred Charollais, Suffolk and Texel lambs. Anim. Sci. 2002, 75, 295–302. [Google Scholar] [CrossRef]
- van der Berg, R.; Levels, F.H.; van der Schee, W. Breed differences in sheep with respect to the accumulation of copper in the liver. Vet. Q. 1983, 5, 26–31. [Google Scholar] [CrossRef]
- Woolliams, J.A.; Woolliams, C.; Suttle, N.F.; Jones, D.G.; Wiener, G. Studies on lambs from lines genetically selected for low and high copper status 2. Incidence of hypocuprosis on improved hill pasture. Anim. Sci. 1986, 43, 303–317. [Google Scholar] [CrossRef]
- Knowles, S.O.; Rounce, J.R.; Grace, N.D.; Lee, J.H. Variation in copper metabolism between two flocks of Romney sheep in response to increasing dietary copper. Proc. New Zealand Soc. Anim. Prod. 1998, 58, 195–198. [Google Scholar]
- Kaler, S.G. Inborn errors of copper metabolism. Handb. Clin. Neurol. 2013, 113, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S. Human copper homeostasis: A network of interconnected pathways. Curr. Opin. Chem. Biol. 2010, 14, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Barry, A.N.; Shinde, U.; Lutsenko, S. Structural organization of human Cu-transporting ATPases: Learning from building blocks. J. Biol. Inorg. Chem. 2010, 15, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.Y.; Gitlin, J.D. Hepatic copper metabolism: Insights from genetic disease. Hepatology 2003, 37, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Horn, N.; Wittung-Stafshede, P. ATP7A-Regulated Enzyme Metalation and Trafficking in the Menkes Disease Puzzle. Biomedicines 2021, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, C.; Aburto, E.M. Review: Animal models of copper-associated liver disease. Comp. Hepatol. 2003, 2, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Kizilaslan, M.; Arzik, Y.; Behrem, S.; Ata, N.; Karaca, O.; Elmaci, C.; Cemal, I. Genome-wide association studies of preweaning growth and in vivo carcass composition traits in Esme sheep. J. Anim. Breed. Genet. 2022, 139, 26–39. [Google Scholar] [CrossRef]
- Lakhssassi, K.; Lahoz, B.; Sarto, P.; Iguácel, L.P.; Folch, J.; Alabart, J.L.; Serrano, M.; Calvo, J.H. Genome-Wide Association Study Demonstrates the Role Played by the CD226 Gene in Rasa Aragonesa Sheep Reproductive Seasonality. Animals 2021, 11, 1171. [Google Scholar] [CrossRef]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef]
- Kijas, J.W.; Townley, D.; Dalrymple, B.P.; Heaton, M.P.; Maddox, J.F.; McGrath, A.; Wilson, P.; Ingersoll, R.G.; McCulloch, R.; McWilliam, S.; et al. A genome wide survey of SNP variation reveals the genetic structure of sheep breeds. PLoS ONE 2009, 4, e4668. [Google Scholar] [CrossRef]
- Ding, R.; Yang, M.; Quan, J.; Li, S.; Zhuang, Z.; Zhou, S.; Zheng, E.; Hong, L.; Li, Z.; Cai, G.; et al. Single-Locus and Multi-Locus Genome-Wide Association Studies for Intramuscular Fat in Duroc Pigs. Front. Genet. 2019, 10, 619. [Google Scholar] [CrossRef]
- Zhao, Y.; Pu, Y.; Liang, B.; Bai, T.; Liu, Y.; Jiang, L.; Ma, Y. A study using single-locus and multi-locus genome-wide association study to identify genes associated with teat number in Hu sheep. Anim. Genet. 2022, 53, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; Vries, J.d.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Prim. 2021, 1, 59. [Google Scholar] [CrossRef]
- Marees, A.T.; Kluiver, H.d.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, e1608. [Google Scholar] [CrossRef]
- Yang, J.; Zaitlen, N.A.; Goddard, M.E.; Visscher, P.M.; Price, A.L. Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 2014, 46, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Li, C.; Fu, Y.; Sun, R.; Wang, Y.; Wang, Q. Single-Locus and Multi-Locus Genome-Wide Association Studies in the Genetic Dissection of Fiber Quality Traits in Upland Cotton (Gossypium hirsutum L.). Front. Plant Sci. 2018, 9, 1083. [Google Scholar] [CrossRef]
- Segura, V.; Vilhjálmsson, B.J.; Platt, A.; Korte, A.; Seren, Ü.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar] [CrossRef]
- Wang, S.-B.; Feng, J.-Y.; Ren, W.-L.; Huang, B.; Zhou, L.; Wen, Y.-J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.-M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Tamba, C.L.; Zhang, Y.-M. A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv 2018, bioRxiv:341784. [Google Scholar] [CrossRef]
- Wen, Y.-J.; Zhang, H.; Ni, Y.-L.; Huang, B.; Zhang, J.; Feng, J.-Y.; Wang, S.-B.; Dunwell, J.M.; Zhang, Y.-M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2017, 18, 906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, J.-Y.; Ni, Y.-L.; Wen, Y.-J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.-M. pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.-L.; Wen, Y.-J.; Dunwell, J.M.; Zhang, Y.-M. pKWmEB: Integration of Kruskal-Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2018, 120, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.L.; Ni, Y.-L.; Zhang, Y.-M. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Öztan, S.; Düring, R.-A. Microwave assisted EDTA extraction-determination of pseudo total contents of distinct trace elements in solid environmental matrices. Talanta 2012, 99, 594–602. [Google Scholar] [CrossRef]
- Gogarten, S.M.; Sofer, T.; Chen, H.; Yu, C.; Brody, J.A.; Thornton, T.A.; Rice, K.M.; Conomos, M.P. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics 2019, 35, 5346–5348. [Google Scholar] [CrossRef]
- Johnson, R.C.; Nelson, G.W.; Troyer, J.L.; Lautenberger, J.A.; Kessing, B.D.; Winkler, C.A.; O’Brien, S.J. Accounting for multiple comparisons in a genome-wide association study (GWAS). BMC Genom. 2010, 11, 724. [Google Scholar] [CrossRef]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C.; et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, X.; Tan, Z.; Ning, C.; Xing, K.; Yang, T.; Pan, Y.; Sun, D.; Wang, C. Genome-Wide Association Study of Piglet Uniformity and Farrowing Interval. Front. Genet. 2017, 8, 194. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.R-project.org/ (accessed on 20 January 2023).
- Yang, J.; Zeng, J.; Goddard, M.E.; Wray, N.R.; Visscher, P.M. Concepts, estimation and interpretation of SNP-based heritability. Nat. Genet. 2017, 49, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Tamba, C.L.; Wen, Y.-J.; Li, P.; Ren, W.-L.; Ni, Y.-L.; Gao, J.; Zhang, Y.-M. mrMLM v4.0.2: An R Platform for Multi-locus Genome-wide Association Studies. Genom. Proteom. Bioinform. 2020, 18, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Hinrichs, A.S.; Kent, W.J. The UCSC Genome Browser. Curr. Protoc. Hum. Genet. 2011, 71, 18.6.1–18.6.33. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.D.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang, W.D.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Rivals, I.; Personnaz, L.; Taing, L.; Potier, M.-C. Enrichment or depletion of a GO category within a class of genes: Which test? Bioinformatics 2007, 23, 401–407. [Google Scholar] [CrossRef]
- Judson, G.J.; Walkley, J.R.W.; James, P.J.; Kleemann, D.O.; Ponzoni, R.W. Genetic variation in trace element status of Merino sheep. Proc. Aust. Soc. Anim. Prod. 1994, 20, 438. [Google Scholar]
- Wang, J.; Fedoseienko, A.; Chen, B.; Burstein, E.; Jia, D.; Billadeau, D.D. Endosomal receptor trafficking: Retromer and beyond. Traffic 2018, 19, 578–590. [Google Scholar] [CrossRef]
- Liu, J.-J. Regulation of dynein-dynactin-driven vesicular transport. Traffic 2017, 18, 336–347. [Google Scholar] [CrossRef]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; Araújo, M.E.G.d.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Pfister, K.K. Distinct functional roles of cytoplasmic dynein defined by the intermediate chain isoforms. Exp. Cell Res. 2015, 334, 54–60. [Google Scholar] [CrossRef] [PubMed]
- McKenney, R.J.; Huynh, W.; Tanenbaum, M.E.; Bhabha, G.; Vale, R.D. Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 2014, 345, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.L.; Song, Y.; Barbar, E. Structural dynamics and multiregion interactions in dynein-dynactin recognition. J. Biol. Chem. 2011, 286, 39349–39359. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.M.; Cater, M.A.; Mercer, J.F.B.; La Fontaine, S. Copper-dependent interaction of dynactin subunit p62 with the N terminus of ATP7B but not ATP7A. J. Biol. Chem. 2006, 281, 14006–14014. [Google Scholar] [CrossRef]
- Polishchuk, E.V.; Concilli, M.; Iacobacci, S.; Chesi, G.; Pastore, N.; Piccolo, P.; Paladino, S.; Baldantoni, D.; van IJzendoorn, S.C.D.; Chan, J.; et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev. Cell 2014, 29, 686–700. [Google Scholar] [CrossRef]
- Chen, K.-E.; Healy, M.D.; Collins, B.M. Towards a molecular understanding of endosomal trafficking by Retromer and Retriever. Traffic 2019, 20, 465–478. [Google Scholar] [CrossRef]
- Lucas, M.; Gershlick, D.C.; Vidaurrazaga, A.; Rojas, A.L.; Bonifacino, J.S.; Hierro, A. Structural Mechanism for Cargo Recognition by the Retromer Complex. Cell 2016, 167, 1623–1635. [Google Scholar] [CrossRef]
- Arredondo, M.; Muñoz, P.; Mura, C.V.; Nùñez, M.T. DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am. J. Physiol.-Cell Physiol. 2003, 284, C1525–C1530. [Google Scholar] [CrossRef]
- Helfer, E.; Harbour, M.E.; Henriot, V.; Lakisic, G.; Sousa-Blin, C.; Volceanov, L.; Seaman, M.N.J.; Gautreau, A. Endosomal recruitment of the WASH complex: Active sequences and mutations impairing interaction with the retromer. Biol. Cell 2013, 105, 191–207. [Google Scholar] [CrossRef]
- Harbour, M.E.; Breusegem, S.Y.; Seaman, M.N.J. Recruitment of the endosomal WASH complex is mediated by the extended ‘tail’ of Fam21 binding to the retromer protein Vps35. Biochem. J. 2012, 442, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Gomez, T.S.; Billadeau, D.D.; Rosen, M.K. Multiple repeat elements within the FAM21 tail link the WASH actin regulatory complex to the retromer. Mol. Biol. Cell 2012, 23, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Krawczak, C.A.; Singla, A.; Starokadomskyy, P.; Deng, Z.; Osborne, D.G.; Li, H.; Dick, C.J.; Gomez, T.S.; Koenecke, M.; Zhang, J.-S.; et al. COMMD1 is linked to the WASH complex and regulates endosomal trafficking of the copper transporter ATP7A. Mol. Biol. Cell 2015, 26, 91–103. [Google Scholar] [CrossRef]
- Materia, S.; Cater, M.A.; Klomp, L.W.J.; Mercer, J.F.B.; La Fontaine, S. Clusterin and COMMD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J. Biol. Chem. 2012, 287, 2485–2499. [Google Scholar] [CrossRef]
- Cox, D.W.; Moore, S.D.P. Copper Transporting P-Type ATPases and Human Disease. J. Bioenergy 2002, 34, 333–338. [Google Scholar] [CrossRef]
- Akram, Z.; Ahmed, I.; Mack, H.; Kaur, R.; Silva, R.C.; Castilho, B.A.; Friant, S.; Sattlegger, E.; Munn, A.L. Yeast as a Model to Understand Actin-Mediated Cellular Functions in Mammals-Illustrated with Four Actin Cytoskeleton Proteins. Cells 2020, 9, 672. [Google Scholar] [CrossRef]
- Zhou, F.-L.; Li, S.C.; Zhu, Y.; Guo, W.-J.; Shao, L.-J.; Nelson, J.; Simpkins, S.; Yang, D.-H.; Liu, Q.; Yashiroda, Y.; et al. Integrating yeast chemical genomics and mammalian cell pathway analysis. Acta Pharmacol. Sin. 2019, 40, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Sowada, N.; Stiller, B.; Kubisch, C. Increased copper toxicity in Saccharomyces cerevisiae lacking VPS35, a component of the retromer and monogenic Parkinson disease gene in humans. Biochem. Biophys. Res. Commun. 2016, 476, 528–533. [Google Scholar] [CrossRef]
- Lei, H.-T.; Mu, X.; Hattne, J.; Gonen, T. A conformational change in the N terminus of SLC38A9 signals mTORC1 activation. Structure 2021, 29, 426–432. [Google Scholar] [CrossRef]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017, 171, 642–654. [Google Scholar] [CrossRef]
- Nnah, I.C.; Wang, B.; Saqcena, C.; Weber, G.F.; Bonder, E.M.; Bagley, D.; Cegli, R.d.; Napolitano, G.; Medina, D.L.; Ballabio, A.; et al. TFEB-driven endocytosis coordinates MTORC1 signaling and autophagy. Autophagy 2019, 15, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Peña-Llopis, S.; Brugarolas, J. TFEB, a novel mTORC1 effector implicated in lysosome biogenesis, endocytosis and autophagy. Cell Cycle 2011, 10, 3987–3988. [Google Scholar] [CrossRef] [PubMed]
- Calvo, J.; Jung, H.; Meloni, G. Copper metallothioneins. IUBMB Life 2017, 69, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Woodman, P. ESCRT-III on endosomes: New functions, new activation pathway. Biochem. J. 2016, 473, e5–e8. [Google Scholar] [CrossRef]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef]
- Tanikawa, S.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Endosomal sorting related protein CHMP2B is localized in Lewy bodies and glial cytoplasmic inclusions in α-synucleinopathy. Neurosci. Lett. 2012, 527, 16–21. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 2015, 60, 47–62. [Google Scholar] [CrossRef]
- Bonora, M.; Bononi, A.; Marchi, E.d.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef]
- Rueda, C.B.; Llorente-Folch, I.; Gonzalez-Sanchez, P.; Contreras, L.; Juaristi, I.; Martinez-Valero, P.; Pardo, B.; Del Arco, A.; Satrustegui, J. Glutamate excitotoxicity and Ca2+-regulation of respiration: Role of the Ca2+ activated mitochondrial transporters (CaMCs). Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, e17–e18. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Amigo, I.; Del Arco, A.; Saheki, T.; Pardo, B.; Satrústegui, J. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef] [PubMed]
- Horn, D.; Barrientos, A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life 2008, 60, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.-J.; Shaki, F.; Ghazi-Khansari, M.; Pourahmad, J. Toxicity of copper on isolated liver mitochondria: Impairment at complexes I, II, and IV leads to increased ROS production. Cell Biochem. Biophys. 2014, 70, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blalchy-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef]
- Anderegg, M.A.; Gyimesi, G.; Ho, T.M.; Hediger, M.A.; Fuster, D.G. The Less Well-Known Little Brothers: The SLC9B/NHA Sodium Proton Exchanger Subfamily-Structure, Function, Regulation and Potential Drug-Target Approaches. Front. Physiol. 2022, 13, 898508. [Google Scholar] [CrossRef] [PubMed]
- Kojima, A.; Toshima, J.Y.; Kanno, C.; Kawata, C.; Toshima, J. Localization and functional requirement of yeast Na+/H+ exchanger, Nhx1p, in the endocytic and protein recycling pathway. Biochim. Biophys. Acta 2012, 1823, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.; Levi, B.P.; Patel, F.I.; Stevens, T.H. The sodium/proton exchanger Nhx1p is required for endosomal protein trafficking in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2000, 11, 4277–4294. [Google Scholar] [CrossRef]
- Karim, M.A.; Brett, C.L. The Na+(K+)/H+ exchanger Nhx1 controls multivesicular body–vacuolar lysosome fusion. Mol. Biol. Cell 2018, 29, 317–325. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Farm | N 1 | Mean (SE) Cu (mg/kg DM) | Min | Max | Mean (SE) Age at Slaughter (Days) | Min | Max | h2(SE) 2 |
|---|---|---|---|---|---|---|---|---|
| 1 | 89 | 159.54 ± 5.22 | 67.67 | 273.42 | 150.76 ± 1.30 | 119 | 165 | 0.67(0.29) |
| 2 | 41 | 79.05 ± 6.80 | 20.57 | 188.28 | 136.78 ± 3.04 | 89 | 172 |
| CHR | SNP 1 | Position | p-Value | Genes 2 (1 Mb) |
|---|---|---|---|---|
| 2 | OAR2_145591151.1 | 136897136 | 5.11 × 10−5 | SLC25A12, DYNC1I2 |
| 2 | s62875.1 | 150905130 | 6.59 × 10−5 | ACVR1C, ACVR1 |
| CHR | SNP | Position (BP) 1 | Genes (1 Mb) 2 | ML-GWAS Methods 3 |
|---|---|---|---|---|
| 1 | s66850.1 | 77025732 | GPR88, ATP5MF | 2, 3, 4, 5 |
| 1 | OAR1_285395930.1 | 263664109 | COL6A1, COL6A2 | 2, 4, 5 |
| 2 | s05644.1 | 14172465 | FRRS1L | 1, 2, 4 |
| 2 | OAR2_145591151.1 * | 136897136 | SLC25A12, DYNC1I2 | 3, 4 |
| 3 | OAR3_132833292.1 | 124516955 | KITLG | 1, 2, 4 |
| 4 | OAR4_77358490.1 | 73042599 | ZNF804B | 2, 3, 5 |
| 6 | s42668.1 | 21789865 | CENPE, SLC9B2, SLC9B2 | 2, 4 |
| 6 | OAR6_47263223.1 | 42315913 | GBA3 | 2, 3, 4, 5 |
| 7 | s25674.1 | 87187746 | NRXN3 | 2, 3, 4 |
| 7 | OAR7_101357352.1 | 93147667 | - | 1, 3, 5 |
| 14 | OAR14_14650208.1 | 14404415 | SPG7, CHMP1A, SHCBP1, VPS35 | 3, 5 |
| 16 | OAR16_25377664_X.1 | 23278766 | IL6ST, SLC38A9 | 2, 4 |
| 23 | OAR23_37101686.1 | 35080540 | GREB1L | 2, 4 |
| Category | Term | Genes | p-Value 1 |
|---|---|---|---|
| GOTERM_MF_DIRECT | GO:0016361~activin receptor activity, type I | ACVR1, ACVR1C | 0.006 |
| GOTERM_CC_DIRECT | GO:0005765~lysosomal membrane | COL6A1, CHMP1A, VPS35, SLC38A9 | 0.007 |
| GOTERM_CC_DIRECT | GO:0048179~activin receptor complex | ACVR1, ACVR1C | 0.008 |
| GOTERM_BP_DIRECT | GO:1902600~hydrogen ion transmembrane transport | SLC9B1, ATP5MF, SLC9B2 | 0.010 |
| GOTERM_CC_DIRECT | GO:0005743~mitochondrial inner membrane | SPG7, SLC25A12, ATP5MF, SLC9B2 | 0.014 |
| GOTERM_CC_DIRECT | GO:0016021~integral component of membrane | ACVR1, KITLG, FRRS1L, GREB1L, NRXN3, SLC38A9, SPG7, IL6ST, SLC25A12, SLC9B1, ATP5MF, SLC9B2 | 0.014 |
| GOTERM_MF_DIRECT | GO:0015385~sodium:proton antiporter activity | SLC9B1, SLC9B2 | 0.015 |
| GOTERM_CC_DIRECT | GO:0030496~midbody | CENPE, CHMP1A, SHCBP1 | 0.016 |
| GOTERM_CC_DIRECT | GO:0005828~kinetochore microtubule | CENPE, CHMP1A | 0.021 |
| GOTERM_BP_DIRECT | GO:0032924~activin receptor signalling pathway | ACVR1, ACVR1C | 0.023 |
| GOTERM_CC_DIRECT | GO:0043235~receptor complex | ACVR1, ACVR1C, IL6ST | 0.023 |
| GOTERM_CC_DIRECT | GO:0097228~sperm principal piece | SLC9B1, SLC9B2 | 0.035 |
| GOTERM_MF_DIRECT | GO:0019838~growth factor binding | ACVR1C, IL6ST | 0.040 |
| GOTERM_MF_DIRECT | GO:0030020~extracellular matrix structural constituent conferring tensile strength | COL6A2, COL6A1 | 0.045 |
| GOTERM_BP_DIRECT | GO:0007080~mitotic metaphase plate congression | CENPE, CHMP1A | 0.047 |
| GOTERM_BP_DIRECT | GO:0001755~neural crest cell migration | ACVR1, KITLG | 0.049 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adeniyi, O.O.; Medugorac, I.; Grochowska, E.; Düring, R.-A.; Lühken, G. Single-Locus and Multi-Locus Genome-Wide Association Studies Identify Genes Associated with Liver Cu Concentration in Merinoland Sheep. Genes 2023, 14, 1053. https://doi.org/10.3390/genes14051053
Adeniyi OO, Medugorac I, Grochowska E, Düring R-A, Lühken G. Single-Locus and Multi-Locus Genome-Wide Association Studies Identify Genes Associated with Liver Cu Concentration in Merinoland Sheep. Genes. 2023; 14(5):1053. https://doi.org/10.3390/genes14051053
Chicago/Turabian StyleAdeniyi, Olusegun O., Ivica Medugorac, Ewa Grochowska, Rolf-Alexander Düring, and Gesine Lühken. 2023. "Single-Locus and Multi-Locus Genome-Wide Association Studies Identify Genes Associated with Liver Cu Concentration in Merinoland Sheep" Genes 14, no. 5: 1053. https://doi.org/10.3390/genes14051053
APA StyleAdeniyi, O. O., Medugorac, I., Grochowska, E., Düring, R. -A., & Lühken, G. (2023). Single-Locus and Multi-Locus Genome-Wide Association Studies Identify Genes Associated with Liver Cu Concentration in Merinoland Sheep. Genes, 14(5), 1053. https://doi.org/10.3390/genes14051053